Xác định nhanh chóng và hiệu quả các ứng viên thuốc sinh học thông qua phân tích sàng lọc thông lượng cao
Hỗ trợ phát triển dược phẩm sinh học bằng công nghệ QTOF và phần mềm Biologics Explorer
Tác giả: Ebru Selen, Zoe Zhang và Kerstin Pohl
SCIEX, Hoa Kỳ
Người biên tập: TS. Lê Sĩ Hưng
Lời giới thiệu: Bài viết này, dịch và tóm tắt phần lớn nội dung chính trong ghi chú kỹ thuật: " Fast and efficient identification of candidate biotherapeutics through high-throughput screening analysis, Link gốc: https://sciex.com/tech-notes/biopharma/fast-and-efficient-identification-of-candidate-biotherapeutics-through-high-throughput-screening-analysis", cung cấp một 1 số kiến thức cơ bản liên quan tới phân tích nguyên vẹn protein và các subunit. Về người biên tập, TS. Lê Sĩ Hưng, tốt nghiệp tiến sĩ tại đại học BOKU Vienna (Cộng hoà Áo) ngành hoá phân tích, đã có trên 10 năm kinh nghiệm làm việc với các thiết bị khối phổ, tập trung vào ứng dụng các kỹ thuật khối phổ trong phân tích các chất chuyển hoá (metabolites) và protein trong các đối tượng mẫu sinh học, ORCID: 0000-0002-0762-3492.
1. Giới thiệu
Intact mass analysis - phân tích khối nguyên vẹn - là kỹ thuật quen thuộc trong phân tích khối lượng phân tử của các phân tử lớn như các protein (ví dụ: myoglobin, bovine serum albumin…), các kháng thể (ví dụ: IgG…), các phân tử có khối lượng lớn (ví dụ: heparin, enoxaparin…) và các tiểu đơn vị (ví dụ: light chain, heavy chain, Fc, Fab…) liên quan. Kỹ thuật này thường sử dụng kết hợp với kỹ thuật sắc ký và khối phổ để xác định khối lượng phân tử của một phân tử sinh học khi phân tử còn nguyên vẹn (có thể chưa biến tính hoặc đã biến tính). Nói cách khác, thay vì phân tích các mảnh hoặc các peptide nhỏ hơn sau khi phân cắt, kỹ thuật này đo khối lượng của toàn bộ phân tử, ví dụ như một protein hoàn chỉnh, một oligonucleotide, hoặc một polymer. Các subunit liên quan, cũng có thể được phân tích khối sau khi tiến hành các phản ứng (ví dụ: cắt, khử, alkyl hoá…) với phân tử hoàn chỉnh. Ví dụ, với chất chuẩn kháng thể đơn dòng thương mại Sigma mAb hoặc các mAb IgG (thường có khối lượng phân tử lớn lên tới 160 KDa, sau khi khử với DTT sẽ thu được 2 chuỗi nặng (heavy chain – Hc, khối lượng phân tử ~ 50-60 KDa) và 2 chuỗi nhẹ (light chain – Lc, khối lượng phân tử ~ 20-30 KDa), các Hc và Lc có thể được coi là các subunit của mAb này. Tương tự khi dùng enzyme thích hợp để cắt (cleave) tại ví trí xác định và khử cầu disulfide sẽ thu được 1 mảnh Fc và 2 mảnh Fab/2, Fc và Fab/2 cũng được coi là các subunit của các mAb này.
Ghi chú kỹ thuật này trình bày một quy trình sàng lọc thông lượng cao sử dụng phân tích khối nguyên vẹn (intact mass analysis) và phân tích khối các tiểu đơn vị (subunit) để xác định các ứng viên thuốc sinh học bằng phần mềm Biologics Explorer kết hợp với hệ thống khối phổ phân giải cao ZenoTOF 7600.
Sắc ký lỏng ghép nối khối phổ (LC-MS) là công cụ tiên phong trong các quy trình phát triển dược phẩm sinh học, cho phép các nhà nghiên cứu xác định, phân tích đặc tính và nghiên cứu hàng chục ứng viên khác nhau để đáp ứng nhu cầu ngày càng tăng về thuốc sinh học. Mặc dù QTOF MS mang lại độ nhạy và độ chính xác khối cao, rất phù hợp cho việc phân tích nguyên vẹn nhiều phân tử lớn và subunit, nhưng việc phân tích dữ liệu thường có nhiều thách thức. Việc phân tích dữ liệu thủ công, chẳng hạn như việc “deconvolute” (giải chập – là thao tác dùng thuật toán để tái tạo lại số khối chính xác của phân tử dựa trên các “charge envelope” trên toàn bộ khoảng phổ) thủ công lần lượt từng file kết quả và đối chiếu với số khối tính toán theo trình tự mong muốn lý thuyết (theoretical sequence information), gây ra những thách thức cho việc sử dụng MS trong sàng lọc ứng viên trong giai đoạn phát triển sớm. Các thách thức này dẫn tới việc sử dụng MS thường được tập trung nhiều hơn vào các giai đoạn phát triển muộn sau đó, sau khi danh sách các thuốc ứng viên đã được thu hẹp bằng các phương pháp khác, cung cấp ít thông tin định tính hơn. Việc có thể xử lý dữ liệu 1 cách tự động, nhanh và tin cậy có thể giúp cải thiện việc đưa ra quyết định chính xác, giảm rủi ro bỏ lọt các ứng viên thuốc sinh học hứa hẹn.
Các thách thức của việc xử lý dữ liệu LC-MS trong sàng lọc ứng viên thuốc ở giai đoạn phát triển sớm có thể được giải quyết bằng phần mềm Biologics Explorer. Một số kháng thể đơn dòng (mAb) và subunit liên quan đã được sử dụng trong nghiên cứu này để đánh giá sự phù hợp liên quan tới khối lượng phân tử của các ứng viên tiềm năng khác nhau. Việc cho phép xử lý đồng thời một số lượng lớn các file dữ liệu, deconvolute tự động và báo cáo trực quan mà không đòi hỏi nhiều sự can thiệp của người dùng là giải pháp cho các thách thức hiện tại. Ngoài ra, tính minh bạch kết quả cao có thể đạt được nhờ tính năng truy cập vào tất cả các bước xử lý dữ liệu khi cần thiết của phần mềm.
2. Các tính năng chính của quy trình sàng lọc protein
Hình 1. Quy trình sàng lọc thuốc sinh học thông lượng cao. Việc sàng lọc khối lượng phân tử (MW) của hàng trăm protein thuốc sinh học khác nhau một cách nhanh chóng và hiệu quả có thể đạt được với quy trình làm việc thân thiện với người dùng nhờ phần mềm Biologics Explorer. Báo cáo cung cấp một cái nhìn tổng quát và trực quan nhanh chóng thể hiện mẫu nào khớp với MW lý thuyết (mong muốn) (dấu tích màu xanh lá cây), những mẫu có sự khác biệt nhỏ về độ chính xác khối (dấu chấm than màu vàng) và những mẫu không phù hợp (dấu x màu đỏ).
3. Phương pháp
Chuẩn bị mẫu: Các ứng viên thuốc sinh học được pha loãng trong đệm Tris 50mM đến nồng độ làm việc là 0,25 μg/μL. Để phân tích các subunit, 150 μL thuốc thử biến tính (denature) protein (guanidine 7,2 M trong đệm Tris 50 mM, pH 7-8) được thêm vào 50 μL mẫu và lắc đều. Dithiothreitol (DTT) được thêm vào (để khử và cắt các cầu disulfide nếu có) đến nồng độ cuối cùng là 100 mM và hỗn hợp được ủ ở 60ºC trong 30 phút. Các mẫu được pha loãng 2 lần với axit formic 0,1% và được chuyển vào các lọ. Đối với phân tích protein nguyên vẹn, mAb ở nồng độ làm việc được chuyển vào các lọ.
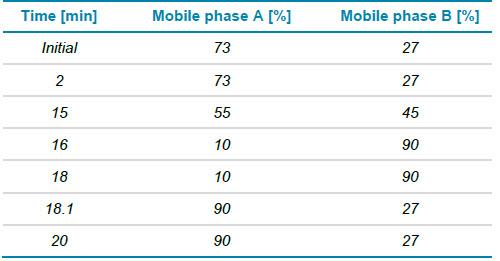
Sắc ký: Tách sắc ký lỏng pha đảo (RPLC) cho protein nguyên vẹn và các subunit được thực hiện với cột Waters BioResolveRP, mAb Polyphenyl, (2,1 mm x 50 mm x 2,7 μm). Tốc độ dòng sử dụng là 0,25 mL/phút, 2 μL mẫu được tiêm lên hệ. Pha động A bao gồm axit formic 0,1% trong nước và pha động B là axit formic 0,1% trong acetonitrile. Bảng 1 thể hiện gradient cho phân tích các protein nguyên vẹn và subunit.
Bảng 1. Gradient phân tích các protein nguyên vẹn và subunit.
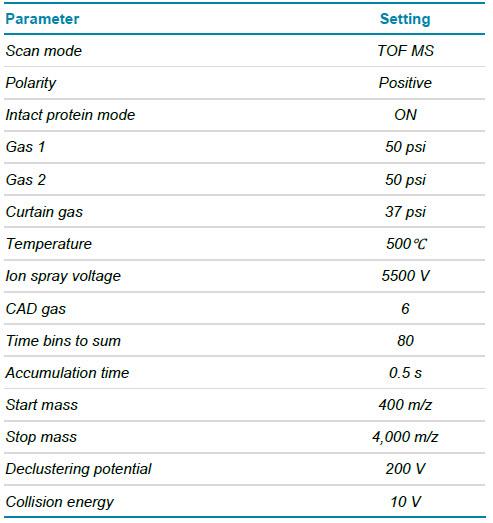
Khối phổ: Dữ liệu được thu thập ở chế độ ion hóa dương TOF MS bằng hệ thống ZenoTOF 7600 với chế độ phân tích protein nguyên vẹn (intact protein mode). Các thông số MS chi tiết được thể hiện trong Bảng 2.
Bảng 2. Các thông số MS để phân tích khối các subunit và protein nguyên vẹn.
Xử lý dữ liệu: Dữ liệu được xử lý và trực quan hóa bằng phần mềm Biologics Explorer phiên bản 1.0.2. Phần mềm Biologics Explorer tương thích với tất cả các máy quang phổ khối chính xác của SCIEX tương thích với dữ liệu ở định dạng .WIFF và .WIFF2.
4. Từ file dữ liệu đến kết quả một cách tin cậy và nhanh chóng
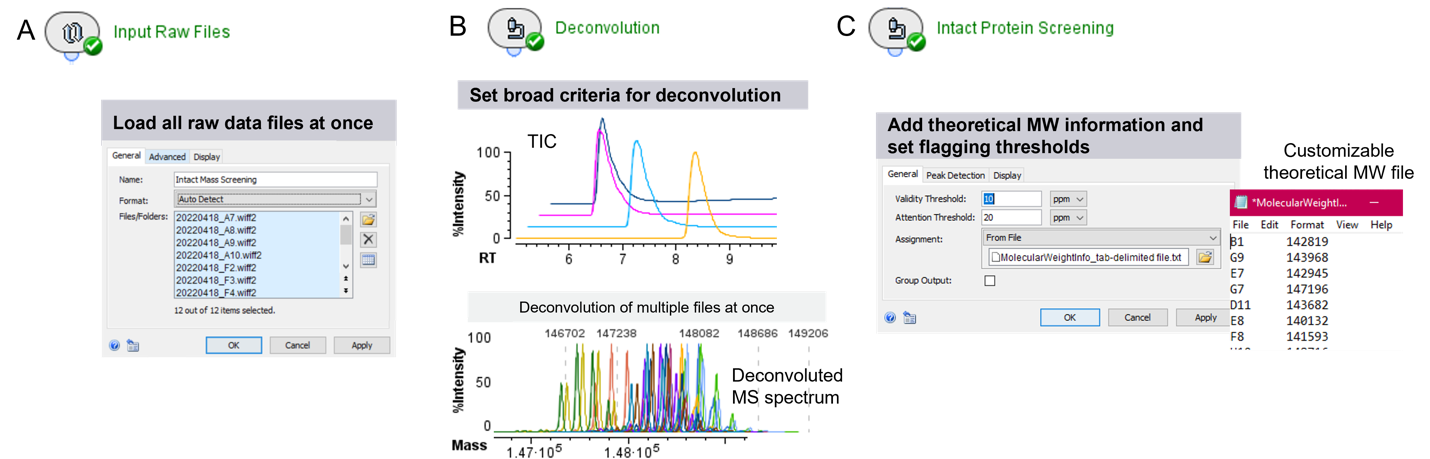
Các mẫu mAb mô phỏng các ứng viên thuốc sinh học khác nhau đã được nạp vào đĩa 96 giếng và dữ liệu TOF MS được thu thập bằng cách sử dụng một phương pháp LC-MS tổng quát (generic LC-MS method). Phương pháp LC-MS tổng quát này cho phép phát hiện nhiều loại mAb khác nhau và các subunit của chúng với chất lượng dữ liệu cao, bỏ qua quá trình tối ưu hóa cụ thể cho từng mẫu để đáp ứng nhu cầu thu thập dữ liệu nhanh chóng và hiệu quả. Để hiểu liệu các protein có được biểu hiện như mong muốn hay không, dữ liệu thô (raw data) TOF MS cần được deconvolute để thu được khối lượng phân tử (MW) của các protein liên quan, sau đó MW đo được này cần phải khớp với MW tính toán từ trình tự protein mong muốn theo lý thuyết. Để bắt đầu quy trình xử lý xác định và đối chiếu MW, tất cả các tệp dữ liệu (mỗi tệp tương ứng với một mẫu protein khác nhau) được nhập vào phần mềm Biologics Explorer cùng một lúc (Hình 2A). Để xác định MW, phạm vi thời gian lưu (RT range) rộng (ví dụ: 5 – 10 phút) được sử dụng để phát hiện píc tự động, deconvolute và tính MW (Hình 2B). Vì các protein khác nhau sẽ có độ kỵ nước (hydrophobicity) khác nhau tùy thuộc vào trình tự (sequence) và sửa đổi (modification) của chúng, nên RT có khả năng bị thay đổi. Do đó, việc phát hiện píc tự động trong một phạm vi RT rộng rất quan trọng để đảm bảo quá trình deconvolute chính xác. Tương tự, MW có thể khác nhau giữa các mẫu protein khác nhau do đó sử dụng một thiết lập khoảng khối khối rộng (ví dụ: 150 Kda – 160 KDa) cho quá trình deconvolute đảm bảo tính linh hoạt cho quy trình làm việc đối với nhiều mẫu khác nhau. Để tự động xác định MW của từng protein ứng viên, phổ MS trong các píc được phát hiện đã được tính trung bình (là thao tác phần mềm trích xuất toàn bộ các phổ MS thuộc cùng một píc từ sắc ký đồ TIC, tổng hợp và tính trung bình về tín hiệu, thành 1 phổ MS duy nhất, ví dụ tính trung bình tất cả phổ MS từ 6,5 tới 7 phút cho píc màu tím trong hình 2B phía trên) và deconvolute cho tất cả các ứng viên cùng một lúc, duy trì tính nhất quán trong quá trình deconvolute (Hình 2B, bên dưới). Việc tham chiếu MW kết quả với MW tính toán từ trình tự lý thuyết được kích hoạt thông qua 1 file text, liên kết đầu ra MW của mỗi tệp dữ liệu với một số khối lý thuyết cụ thể (ví dụ: mẫu trong giếng B1 có số khối tính toán theo lý thuyết là 142.819 Da) (Hình 2C). Ngoài ra, 2 ngưỡng để đánh dấu kết quả (flagging) có thể được thiết lập dựa trên sự khác biệt nhỏ và đáng kể giữa số khối đo được và số khối mong đợi (lý thuyết).
Hình 2. Sàng lọc một nhóm thuốc sinh học bằng phần mềm Biologics Explorer. Người dùng tải lên các file kết quả thu được sau khi đo bằng ZenoTRAP 7600 (A). Các píc được tự động phát hiện từ sắc ký đồ TIC (B, bảng trên), dữ liệu được deconvolute theo lô (B, bên dưới) bằng cách sử dụng 1 cài đặt chung và tổng quát. Số khối của các protein nguyên vẹn và các subunit đo được và được tự động sàng lọc dựa trên file text chứa các giá trị MW mục tiêu. Ngoài ra, có thể đặt ngưỡng đánh dấu kết quả dựa trên độ lệch khối của MW đo được so với MW lý thuyết cho báo cáo (C).
Các phương pháp sàng lọc hiện tại cho các protein nguyên vẹn và subunit đang gặp khó khăn do sự thiếu tự động hóa trong phân tích dữ liệu. Mặc dù vẫn có thể deconvolute nhiều file dữ liệu đồng thời với hầu hết các phần mềm trên thị trường, người dùng vẫn cần kiểm tra thủ công các kết quả để xác định sự phù hợp giữa các MW tính theo chuỗi (ví dụ: chuỗi peptide trong trường hợp của protein, hoặc chuỗi polymer…) lý thuyết và MW đo được sau khi đã deconvolute (thực nghiệm). Trong một môi trường có nhiều áp lực về thời gian, luôn mong muốn sàng lọc hàng trăm ứng viên mỗi ngày, việc thường quy hoá các quy trình trở nên cần thiết nhưng cũng đầy thách thức. Thách thức này được giải quyết bằng một phương pháp phân tích tổng quát, liên kết MW lý thuyết với dữ liệu thực nghiệm một cách hiệu quả. Liên kết tự động được khởi tạo bởi một file text được phân tách bằng dấu tab bao gồm tên file mẫu và thông tin MW lý thuyết (Hình 2C). Phần mềm tự động tạo liên kết giữa thông tin liên quan tới MW lý thuyết và dữ liệu thực nghiệm bằng cách sử dụng tên của file mẫu. Sự khác biệt giữa số khối lý thuyết và số khối đo được từ thực nghiệm, được thể hiện ở đơn vị ppm, và được so sánh với các ngưỡng thẩm định (validity thresshold) và cần chú ý (attention thressholds) (Ví dụ: validity thresshold = 10 ppm và attention thresshold = 20 ppm), giúp việc kiểm tra kết quả vô cùng dễ dàng và trực quan (Hình 1 và 3). Hình 3 thể hiện các ví dụ về báo cáo mẫu ở định dạng PDF cho phân tích protein nguyên vẹn và subunit của các ứng viên thuốc sinh học. Một ứng viên sẽ được liên kết với 1 trong 3 tùy chọn: hợp lệ (valid) với dấu kiểm màu xanh lá cây nếu sự khác biệt giữa số khối mong đợi (expected mass) và số khối đo được (detected mass) thấp hơn ngưỡng hợp lệ tuyệt đối (ví dụ: ≤ 10 ppm), cần cẩn thận (critical) với dấu chấm than màu vàng khi sự khác biệt thấp hơn ngưỡng cần chú ý nhưng cao hơn ngưỡng hợp lệ (ví dụ: > 10 ppm nhưng ≤ 20 ppm), hoặc không hợp lệ với dấu gạch chéo màu đỏ nếu sự khác biệt cao hơn ngưỡng cần chú ý (ví dụ: > 20 ppm), cho thấy cần phải kiểm tra lại kết quả.
Hình 3. Kết quả có thể được truy cập chỉ với một nút duy nhất giúp tăng hiệu quả trong việc đưa ra quyết định trong môi trường có áp lực về thời gian. Kết quả được cung cấp ở định dạng pdf có thể tùy chỉnh với thông tin về tên mẫu, số khối dự kiến (expected mass) và số khối đo được (detected mass), sai số về số khối và diễn giải trực quan (valid) để truy vấn kết quả hiệu quả. Ví dụ cho thấy kết quả thu được từ phân tích số khối nguyên vẹn ở trên cùng (intact) và kết quả chuỗi nhẹ (Lc) và nặng (Hc) từ phân tích subunit ở dưới.
Việc diễn giải trực quan các kết quả bằng các biểu tượng trực quan làm tăng hiệu quả trong việc đưa ra quyết định trong môi trường chịu nhiều áp lực về thời gian. Người dùng có thể điều chỉnh cài đặt các ngưỡng thẩm định và cần chú ý để phù hợp với từng nhu cầu của mình (Hình 3). Ngoài tên tệp, thông tin liên quan về số khối dự kiến và số khối đo được cũng như độ lệch ppm được hiển thị. Hơn nữa, thông tin trong báo cáo có thể tùy chỉnh theo các mẫu.
5. Tính minh bạch trong phân tích dữ liệu và kết quả
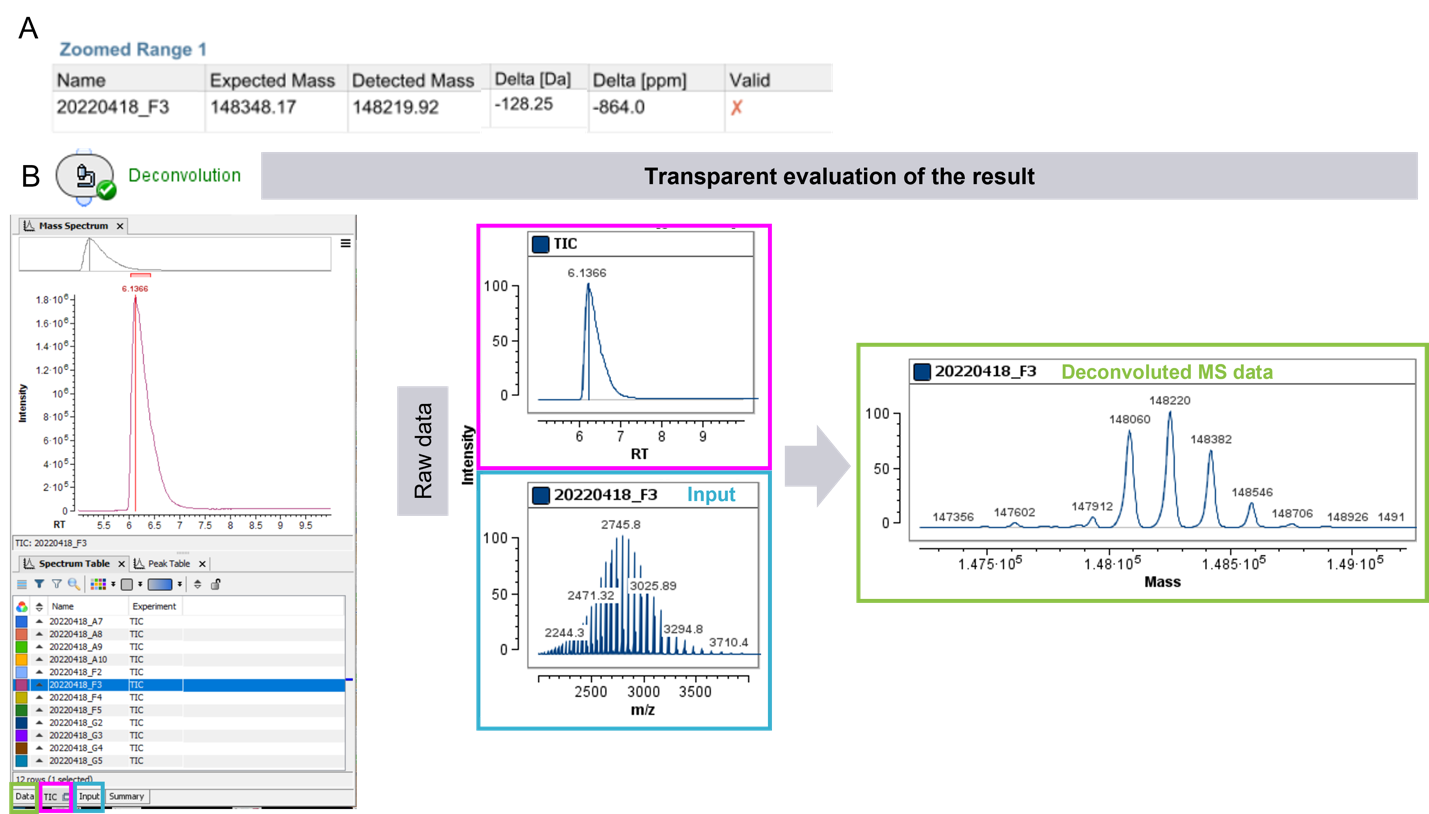
Quy trình làm việc thông lượng cao được tối ưu hóa để tăng tốc quá trình sàng lọc và ra quyết định sử dụng giao diện thân thiện với người dùng cùng khả năng báo cáo/hiển thị kết quả trực quan thông minh. Để kiểm tra một mẫu bất kỳ, nhất là với các mẫu có độ độ lệch (khối) đáng kể so với mong đợi, phần mềm cũng cung cấp tùy chọn để truy vấn dữ liệu ở mỗi bước xử lý nhằm đảm bảo đầy đủ tính minh bạch. Ví dụ, dữ liệu cho mẫu trong giếng F3 đã được kiểm tra (Hình 4). Dấu màu đỏ từ báo cáo sàng lọc nhanh cho mẫu F3 (Hình 3 và Hình 4A) thể hiện rằng mẫu F3 cần được kiểm tra thêm. Việc kiểm tra nhanh bảng trong báo cáo cho thấy sự khác biệt là ~128 Da giữa số khối dự kiến và số khối đo được cho mẫu F3. Dữ liệu thô và dữ liệu đã deconvolute đại diện sau đó đã được truy cập lại để kiểm tra chất lượng dữ liệu (Hình 4B): Một píc có giá trị S/N rất tốt trong TIC cho thấy việc bơm mẫu vào hệ thống LC-MS thành công, và kết quả thu được tốt không bị ảnh hưởng bởi nhiễu nền.
Hình 4. Minh họa về kiểm tra dữ liệu minh bạch trong phần mềm Biologics Explorer. Mẫu F3 cho thấy độ lệch khối cao hơn ngưỡng cần chú ý trong báo cáo > 20 ppm (A). Người dùng có thể kiểm tra dữ liệu thô gồm TIC, phổ TOF MS thu được và dữ liệu đã deconvolute (B) để kiểm tra.
Hơn nữa, với mẫu FG3 một nhóm trạng thái điện tích của protein (charge state envelope) với sự phân tách đường nền (baseline separation) của các dạng protein (proteoform) được thể hiện rõ ràng. Dữ liệu deconvolute thu được cũng cho thấy một kiểu dạng mAb chung (common pattern), với các proteform cách nhau 160 Da, 162 Da… có thể tương ứng với các phân tử đường trong chuỗi glycan gắn vào mAb. Giải thích cho sự không khớp giữa khối lượng được deconvolute mong đợi và đo được có thể là do sự mất 1 axit amin (lysine) trong quá trình phản ứng, dẫn đến MW đo được thấp hơn so với MW mong đợi. Tại bước này, có thể đưa ra quyết định tiếp tục kiểm tra mẫu hay tập trung vào các mẫu đã vượt qua các tiêu chí. Để điều tra thêm về sự mất axit amin này, có thể thực hiện phân tích sâu hơn bằng việc lập bản đồ peptide (peptide mapping) và có thể điều tra sự sai khác trình tự và (các) sửa đổi của thuốc sinh học ứng viên.
6. Kết luận
Việc phát triển dược phẩm sinh học có thể được tăng tốc bằng cách sàng lọc nhanh chóng và hiệu quả sử dụng kỹ thuật phân tích khối các protein nguyên vẹn và các subunit liên quan nhờ LC-QTOF kết hợp với phần mềm Biologics Explorer. Quy trình làm việc thân thiện với người dùng giúp thu hẹp các khoảng trống phân tích dữ liệu hiện tại, đồng thời cho phép trải nghiệm phân tích dữ liệu minh bạch.
Đội ngũ chuyên gia của Thăng Long sẵn sàn hỗ trợ bạn lựa chọn giải pháp phù hợp
12344