Phương pháp phân tích các chất hướng thần mới trong dược học và độc học - Chương 3
Methods for Novel Psychoactive Substance Analysis, Methods in Pharmacology and Toxicology
Biên soạn bởi
Marta Concheiro
Department of Sciences, John Jay College of Criminal Justice, City University of New York,
New York, NY, USA
Karl B. Scheidweiler
Scientific Affairs, Toxicology R&D, Abbot/Immunalysis, Pomona, CA, USA
Chương 3: Phân Tích Mục Tiêu Các Chất Phenethylamin, Tryptamin và Piperazin trong Máu và Nước Tiểu
Tác giả: Steven Towler và Marta Concheiro
Lời giới thiệu: Sau đây là bản tóm tắt lược dịch chương 3 từ cuốn sách: “Methods for Novel Psychoactive Substance Analysis, Methods in Pharmacology and Toxicology, của các biên tập Marta Concheiro và Karl B. Scheidweiler; DOI: https://doi.org/10.1007/978-1-0716-2605-4_3”. Người dịch, TS. Lê Sĩ Hưng, tốt nghiệp tiến sĩ tại đại học BOKU Vienna (Cộng hoà Áo) ngành hoá phân tích, đã có trên 10 năm kinh nghiệm làm việc với các thiết bị khối phổ, tập trung vào ứng dụng các kỹ thuật khối phổ trong phân tích các chất chuyển hoá (metabolites) và protein trong các đối tượng mẫu sinh học, ORCID: 0000-0002-0762-3492. Chương 3 cuốn sách này cung cấp một cái nhìn tổng quan về các phương pháp phân tích hiện tại để phát hiện và định lượng các chất gây ảo giác và kích thích như phenethylamin, tryptamin và piperazin trong các mẫu sinh học như máu, huyết tương, huyết thanh và nước tiểu. Các chất này, bao gồm các chất gây ảo giác cổ điển như LSD và các hợp chất mới nổi như NBOMe, có các đặc tính hóa lý tương tự, gây khó khăn cho việc phân tích.
Tóm tắt
Phenethylamin, tryptamin và piperazin là một nhóm phức tạp các chất gây ảo giác và kích thích. Việc phân tích các chất này trong các phòng thí nghiệm độc chất có thể không được thực hiện thường xuyên và trở thành một thách thức do số lượng lớn của các chất này, sự xuất hiện liên tục của các dẫn xuất mới trên thị trường và nồng độ thấp trong các mẫu sinh học do hiệu lực cao của chúng. Chương này cung cấp tóm tắt về các phương pháp hiện tại có sẵn để phân tích mục tiêu các chất này trong huyết tương, huyết thanh, máu và mẫu nước tiểu. Tổng quan quan trọng này tập trung chủ yếu vào các phương pháp xác nhận được công bố gần đây và làm nổi bật các khía cạnh thực tế của các phương pháp này, chẳng hạn như các chất phân tích mục tiêu được khuyến nghị, nồng độ dự kiến, độ ổn định của các hợp chất này trong các mẫu máu và nước tiểu, và các khía cạnh phân tích, bao gồm cả quá trình thủy phân mẫu, quy trình chiết và phân tích bằng dụng cụ.
Từ khóa: Phenethylamines, Tryptamines, Piperazines, Huyết thanh, Huyết tương, Máu, Nước tiểu, GC-MS, LC-MS
1. Giới thiệu
Phenethylamin và tryptamin là các chất gây ảo giác mạnh tạo ra tác dụng gây ảo giác của chúng bằng cách liên kết với các thụ thể serotonin, 5-HT2A và các thụ thể khác. Thụ thể serotonin 5-HT2A có liên quan đến các hành vi phức tạp bao gồm trí nhớ làm việc, quá trình nhận thức và các rối loạn cảm xúc như tâm thần phân liệt [1]. Cụ thể, phenethylamin có ái lực cao với các thụ thể serotonin 5-HT2A, cũng như sự chủ vận một phần (partial agonism – Chú thích của người dịch: chủ vận là một chất hóa học có khả năng liên kết với một thụ thể cụ thể trong tế bào và kích hoạt thụ thể đó, tạo ra một phản ứng sinh học) tại thụ thể 5-HT2c [2]. Mặt khác, tryptamin hoạt động như chất chủ vận của các thụ thể serotonin 5-HT2A và 5-HT1A.
Phenethylamin có liên quan về mặt hóa học đến alkaloid gây ảo giác tự nhiên mescaline. Một số phenethylamin gây ảo giác đã được Shulgin tổng hợp trong những năm 1970 và 1980 [3]. Các chất này, cũng như các dẫn xuất mới, đã xuất hiện trở lại và xuất hiện trên thị trường kể từ những năm 2000 [2, 4]. Các thành viên của lớp chất gây ảo giác này có thể được chia nhỏ hơn về mặt hóa học thành ba phân lớp: 2C, 2C-T và chuỗi DO (xem Hình 1, 2, 3 và 4). Các chuỗi 2C là dimethoxyphenethylethanamine chứa hai nhóm methoxy ở vị trí 2 và 5 của vòng benzen và hai carbon (2C) giữa amin và vòng benzen. Trong họ 2C, các dẫn xuất mới thuộc nhóm NBOMe (N-2-methoxybenzyl phenethylamin). Lớp thuốc NBOMe bao gồm 25B-, 25C-, 25D-, 25H-, 25I- và 25 T2-NBOMe. Trong nhóm này, việc bổ sung một chất thay thế N-benzyl vào các chất gây ảo giác phenethylamin làm tăng đáng kể ái lực của thụ thể 5-HT2A [5]. Các chuỗi 2C-T là dimethoxyphenethylethanamine thường chứa một nhóm thio-alkyl hóa ở vị trí 4 của vòng. Chuỗi DO là dimethoxyphenylpropanamine chứa một methyl trên chuỗi aminoethyl và một halogen hoặc nhóm alkyl ở vị trí 4 của vòng benzen. Chuỗi DO tạo ra các tác dụng gây ảo giác mạnh hơn so với chuỗi 2C hoặc 2C-T [6]. Trong khi một số phenethylamin được liệt kê trong Danh mục I và II của Công ước Liên Hợp Quốc năm 1971 về các chất gây hướng thần, hầu hết các chất mới không thuộc sự kiểm soát quốc tế. Ví dụ, ở Hoa Kỳ, hầu hết các hóa chất 2C-x đều thuộc Danh mục I theo Đạo luật An toàn và Đổi mới của Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ năm 2012 và Đạo luật Tương tự Liên bang [7], và NBOMe, 25I-NBOMe, 25B-NBOMe và 25C-NBOMe hiện là các chất thuộc Danh mục I theo 21 CFR 1308.11 (d) [8].
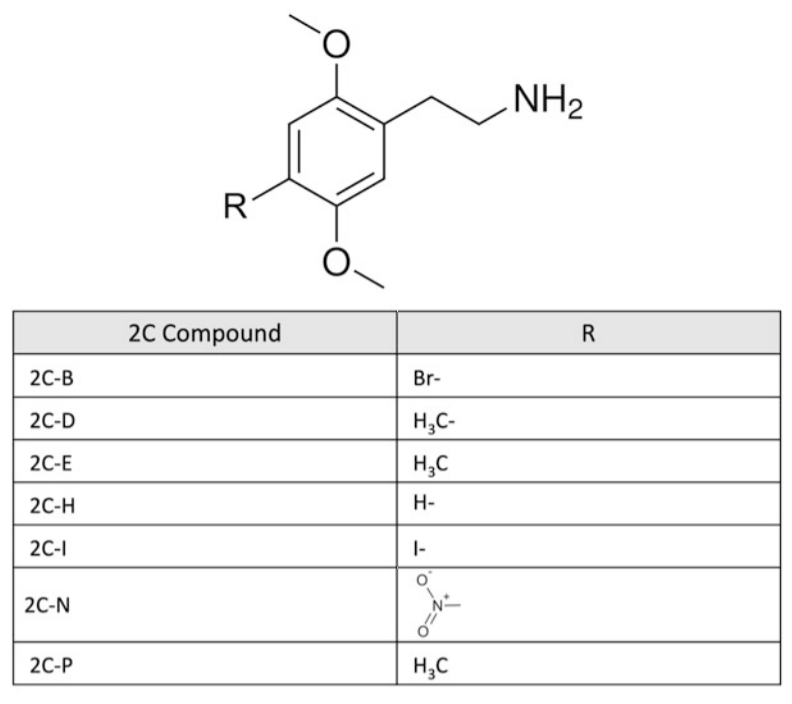
Hình 1. Cấu trúc hóa học của các hợp chất 2C (phenethylamin)
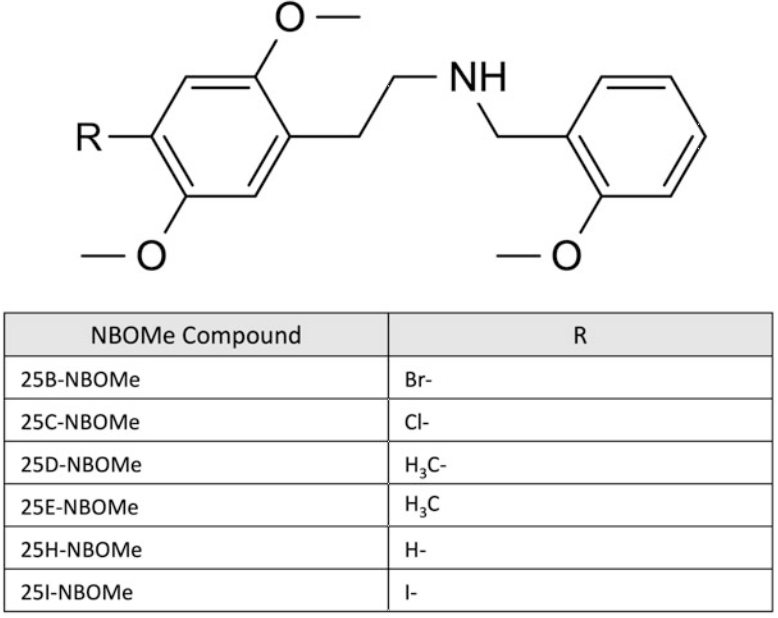
Hình 2. Cấu trúc hóa học của các hợp chất NBOMe chọn lọc (phenethylamin)
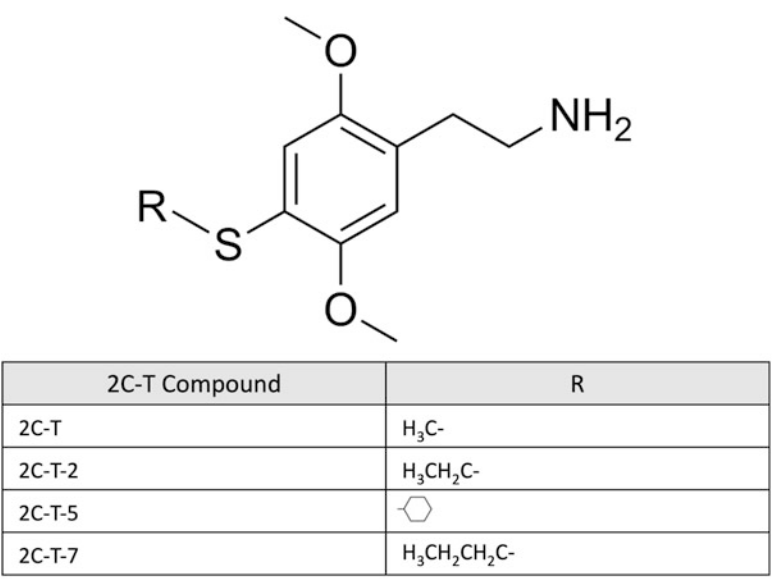
Hình 3. Cấu trúc hóa học của các hợp chất 2C-T chọn lọc (phenethylamin)
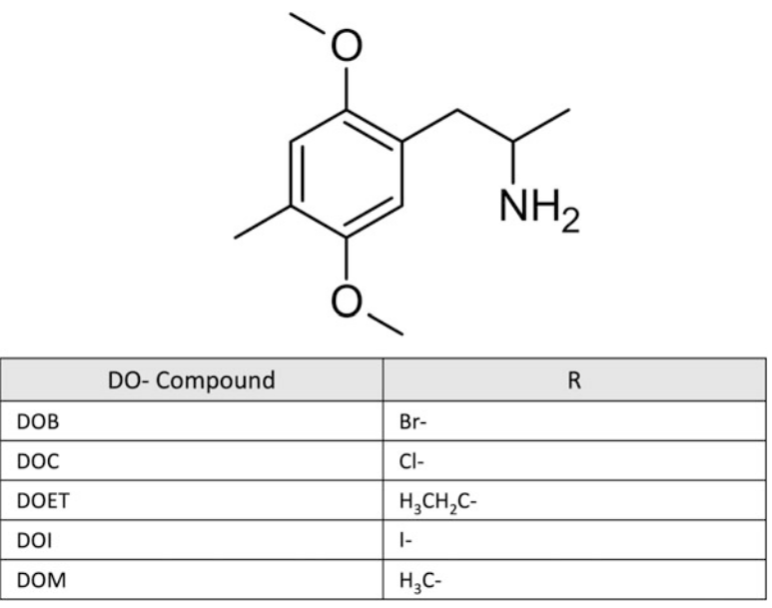
Hình 4. Cấu trúc hóa học của các hợp chất DO chọn lọc (phenethylamin)
Các tryptamin được biết đến nhiều là psilocin, psilocybin, N,N-dimethyltryptamine (DMT) và lysergic acid diethylamide (LSD). Về mặt hóa học, chúng có liên quan đến chất dẫn truyền thần kinh serotonin. Việc sử dụng psilocybin tự nhiên trở nên phổ biến vào cuối những năm 1950 ở Hoa Kỳ, trong khi các tryptamin tổng hợp xuất hiện trên thị trường ma túy bất hợp pháp trong suốt những năm 1990. Gần đây, các chất gây ảo giác tryptamin mới nổi là alpha-methyltryptamine (AMT), 5-methoxy-N,N-dimethyltryptamine (5-MeO-DMT), và 5-methoxy-N,N-diisopropyltryptamine (5-MeO-DIPT). Trong số các dẫn xuất LSD mới xuất hiện trên thị trường gần đây bao gồm 1-propionyl-LSD (1P-LSD), lysergic acid 2,4-dimethylazetidide (LSZ), N-allyl-nor-LSD (AL-LAD), N-ethyl-nor-LSD (ETH-LAD), lysergic acid morpholide (LSM-775), N-ethyl-N-cyclopropyllysergamide (ECPLA) và 1-propionyl-ETH-LAD (1P-ETH-LAD) [9]. Xem Hình 5 và 6. Psilocin và psilocybin là các tryptamin duy nhất thuộc sự kiểm soát quốc tế (được liệt kê trong Danh mục I của Công ước năm 1971), trong khi một số chất khác được kiểm soát ở cấp quốc gia ở một số quốc gia.
Nhóm chất thứ ba bao gồm trong chương này, piperazin, là các thuốc kích thích tâm thần chứa vòng piperazin và tạo ra các tác dụng như 3,4-methylenedioxymethamphetamine (MDMA), amphetamine và methamphetamine (xem Hình 7 và 8). Piperazin được chia về mặt hóa học thành hai loại, cụ thể là benzylpiperazin (xem Hình 7), như 1-benzylpiperazine (BZP), 1-(3,4-methylenedioxybenzyl) piperazine (MDPP) và 1,4-dibenzylpiperazine (DBZP), và phenylpiperazin (xem Hình 8), như 1-(3-tri-fluoromethylphenyl) piperazine (TFMPP), 1-(4-fluorophenyl) piperazine (pFPP), và 1-(3-chlorophenyl) piperazine (mCPP) [10]. Theo Văn phòng Liên hợp quốc về Ma túy và Tội phạm (UNODC), bản thân BZP ban đầu được phát triển như một loại thuốc chống trầm cảm tiềm năng nhưng được phát hiện có các đặc tính tương tự như amphetamine và do đó dễ bị lạm dụng. Vào cuối những năm 1990, BZP nổi lên ở New Zealand như một "lựa chọn thay thế hợp pháp" cho MDMA và methamphetamine. Ở Châu Âu, việc sử dụng BZP lần đầu tiên được báo cáo ở Thụy Điển vào năm 1999, nhưng chỉ trở nên phổ biến như một NPS từ năm 2004 trở đi cho đến khi các biện pháp kiểm soát chất này được đưa ra vào năm 2008. Cả BZP và bất kỳ piperazin nào khác đều không thuộc sự kiểm soát quốc tế, mặc dù một số quốc gia đã đưa ra các biện pháp kiểm soát quốc gia đối với piperazin.
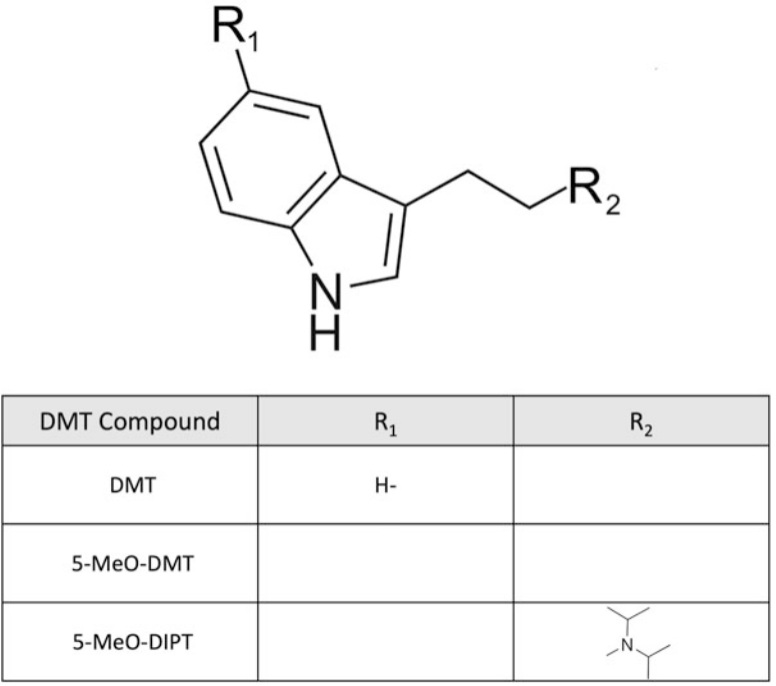
Hình 5. Cấu trúc hóa học của N,N-dimethyltryptamine (DMT) và các dẫn xuất mới (tryptamines)
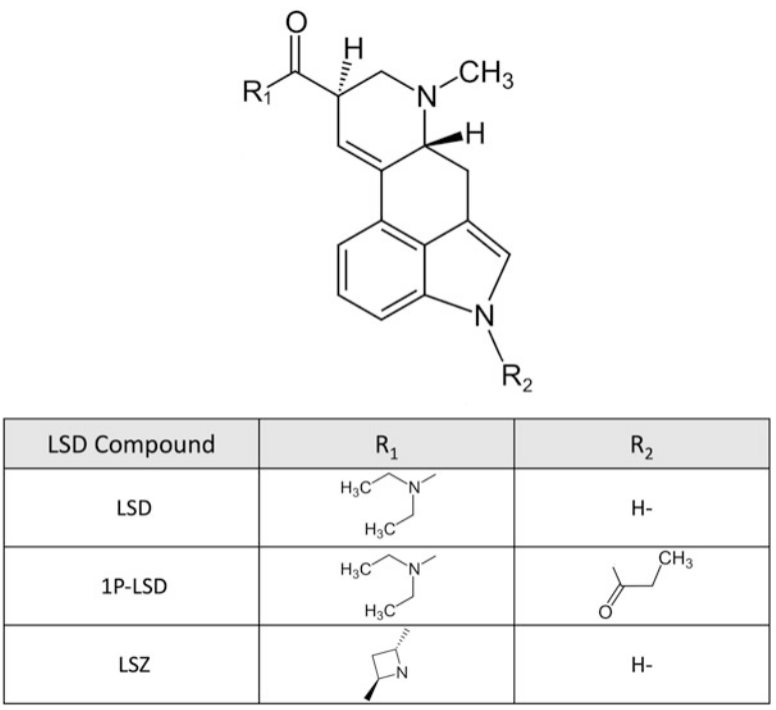
Hình 6. Cấu trúc hóa học của lysergic acid diethylamide (LSD) và các dẫn xuất mới (tryptamines)
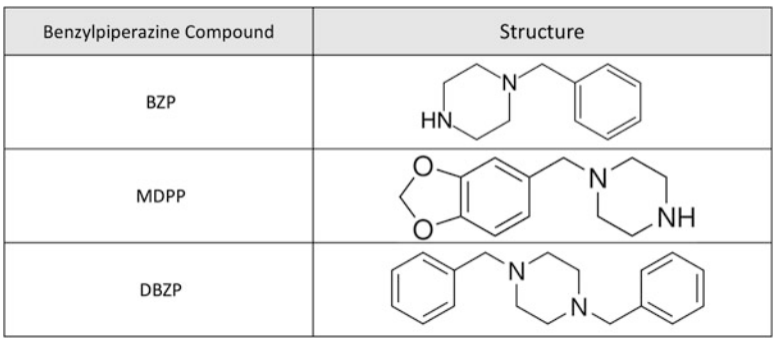
Hình 7. Cấu trúc hóa học của các hợp chất benzylpiperazin chọn lọc (piperazines)
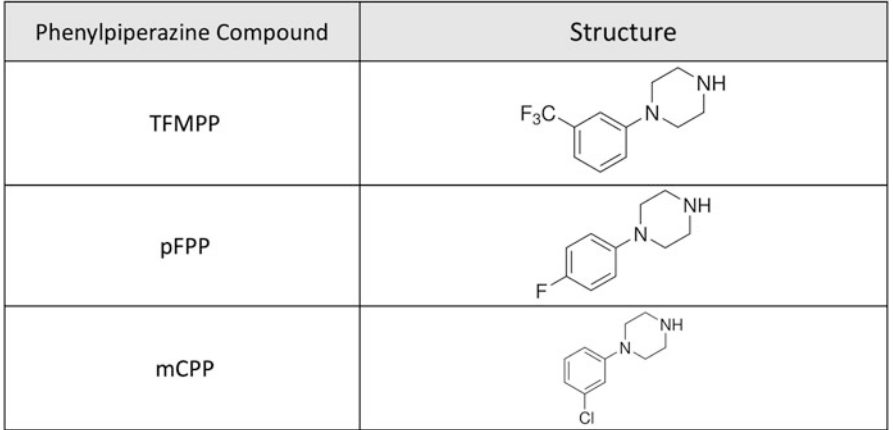
Hình 8. Cấu trúc hóa học của các hợp chất phenylpiperazin chọn lọc (piperazines)
Phenethylamin, piperazin và tryptamin, mặc dù thuộc các nhóm thuốc khác nhau, nhưng tất cả đều có các đặc tính hóa lý tương tự. Phenethylamin, piperazin và tryptamin là các phân tử hữu cơ nhỏ (khối lượng phân tử nhỏ hơn 500 Da), có tính bazơ với pKa từ 8 đến 11 và có tính ưa lipit thấp, log P nhỏ hơn 5 (Bảng 1). Các đặc tính hóa lý này là chìa khóa để thiết kế các quy trình chiết từ chất nền sinh học và cho các phương pháp phát hiện của chúng bằng các kỹ thuật phân tích khác nhau.
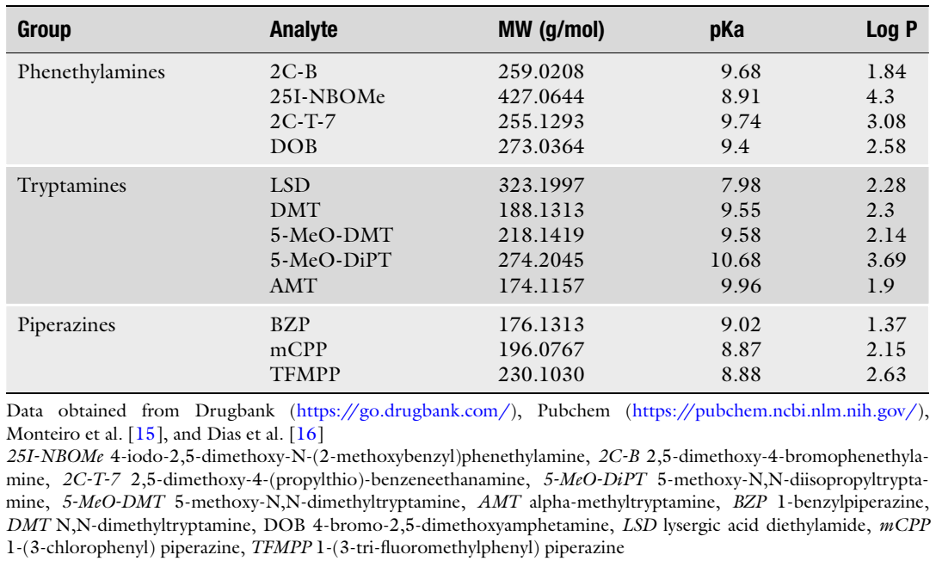
Bảng 1. Khối lượng phân tử đơn đồng vị (MW, g/mol ), pKa và Log P của các phenethylamin, tryptamine và piperazines được lựa chọn
Chương này, cung cấp các hướng dẫn thực tế để phân tích định lượng phenethylamin, tryptamin và piperazin trong máu, huyết thanh, huyết tương và nước tiểu, bao gồm tổng quan về các phương pháp đã công bố, làm nổi bật các chất phân tích mục tiêu được khuyến nghị, nồng độ dự kiến và các kỹ thuật chiết mẫu phổ biến nhất, kỹ thuật tách sắc ký phân tích và phát hiện.
2 Xác định các phenethylamin trong máu và nước tiểu
Bảng 2 tóm tắt các đặc điểm chính của các phương pháp phân tích được chọn để xác định phenethylamin, bao gồm các chuỗi 2C-, 2C-T- và DO-, trong máu toàn phần, huyết tương, huyết thanh và nước tiểu. Tất cả các phương pháp này cho thấy độ nhạy và độ đặc hiệu cao và đã được xác thực đầy đủ như các phương pháp định lượng, và hầu hết trong số đó đã được áp dụng cho các mẫu thực.
Bảng 2. Các phương pháp phân tích xác định phenethylamin trong máu toàn phần, huyết tương, huyết thanh và nước tiểu
2.1 Các chất phân tích mục tiêu cho các phenethylamin
Tất cả các phương pháp này đều có các chất gốc là mục tiêu phân tíchcho bất cứ loại mẫu sinh học nào được phân tích. Đây là một cách tiếp cận phổ biến với loại chất này vì dữ liệu hạn chế về quá trình trao đổi chất của chúng ở người và/hoặc các chất chuyển hóa không có sẵn trên thị trường [6]. Pichini et al. [21], sử dụng quy trình được tóm tắt trong Bảng 5, đã phân tích các mẫu nước tiểu đã thủy phân so với chưa thủy phân và báo cáo nồng độ các hợp chất 2C cao hơn trong các mẫu đã thủy phân. Điều này chỉ ra rằng các hợp chất này có thể được tìm thấy trong nước tiểu dưới dạng các liên hợp glucuronide và/hoặc sulfat. Trong đó, các mẫu nước tiểu được thủy phân bằng β-glucuronidase từ Helix pomatia (114.400 đơn vị β-glucuronidase/mL; 3290 đơn vị sulfatase/mL), 16 giờ ở 37 ° C.
2.2 Liều lượng và nồng độ của các phenethylamin
Do hiệu lực cao của chúng, các liều phenethylamin thường được báo cáo là thấp. Đối với các thuốc 2C, liều lượng dao động từ 6 đến 60 mg, đối với các hợp chất 2C-T từ 8–30 mg, và các hợp chất DO từ 1–10 mg, và đối với dẫn xuất NBOMe, liều lượng nằm trong phạm vi µg (500-800 µg) [3, 6, 22–24]. Do đó, nồng độ các chất này trong máu và nước tiểu dự kiến cũng thấp (pg/mL và ng/mL thấp). Ví dụ, việc sử dụng các chất gây ảo giác 2C dẫn đến nồng độ trong huyết tương trong khoảng 27,3–43 ng/mL [17, 25], và việc sử dụng NBOMe dẫn đến nồng độ trong máu trong khoảng 0,034 – 2,78 ng/mL [1, 19, 23]. Các phương pháp phân tích được tóm tắt trong Bảng 2 cho phép đạt được giới hạn định lượng (LOQ) từ 0,03 đến 10 ng/mL trong máu, huyết tương, huyết thanh và nước tiểu.
2.3 Độ ổn định của các phenethylamin
Một yếu tố quan trọng trước phân tích cần được xem xét trong việc phân tích các phenethylamin là độ ổn định của chúng trong các mẫu sinh học. Một số nghiên cứu từ Bảng 2 đã đánh giá độ ổn định trong các mẫu tăng cường nhưng không phải trong các mẫu thực (stability in fortified but not in authentic samples). Caspar et al. [18] nhận thấy có sự thay đổi xảy ra đối với 2C-N, 2C-B-fly và 2C-E sau các chu kỳ đông lạnh-rã đông, cũng như 2C-E và NBOMe ở nhiệt độ phòng trong huyết tương. Habrdova et al. [4] cũng quan sát thấy các vấn đề về sự ổn định của 2C-I và 2C-D trong huyết tương sau các chu kỳ đông lạnh-rã đông. Cunha et al. [26] cũng báo cáo các vấn đề về độ ổn định với các chất NBOMe trong máu toàn phần. Ở nhiệt độ phòng, 25B-, 25C-, 25D-, 25E-, 25G-, 25H- và 25I-NBOMe cho thấy độ ổn định kém sau 15 ngày và giảm hơn 20% ở 4°C sau 180 ngày cho hầu hết các chất so với nồng độ ban đầu. Ở -20°C, tất cả các chất phân tích đều ổn định trong 180 ngày. Mặt khác, Curtis et al. [27] đã báo cáo độ ổn định tốt của 2C-T-7 lên đến 70 ngày trong máu bảo quản bằng natri florua ở 4°C. Poklis et al. [19] đã báo cáo các dẫn xuất NBOMe ổn định ở trạng thái đông lạnh (1 tháng) hoặc nhiệt độ phòng (72 giờ) trong nước tiểu. Ngoại lệ duy nhất là 2CT-NBOMe có độ lệch 232–243% so với nồng độ mục tiêu của các mẫu kiểm soát chất lượng được bảo quản ở nhiệt độ phòng trong 72 giờ. Do các vấn đề về độ ổn định này và nồng độ thấp có trong các mẫu sinh học, các mẫu để phân tích phenethylamin nên được bảo quản đông lạnh cho đến khi phân tích.
2.4 Các chất nội chuẩn sử dụng cho phân tích các phenethylamin
Hầu hết các phương pháp đã công bố đều sử dụng các chất tương tự được deuteri hóa của các hợp chất có liên quan về mặt hóa học, ngoại trừ Poklis et al. đã sử dụng chất tương tự được deuteri hóa của một trong các chất phân tích mục tiêu (25I-NBOMe-d3) [19]. Các chất nội chuẩn này được sử dụng là trimipramine-d3 [18], MDPV-d8 [20], amphetamine-d5, methamphetamine-d5, MDA-d5, MDMA-d5, MDEA-d5 [4] và mescaline-d9 [4, 6].
2.5 Quy trình chiết các phenethylamin
Do bản chất bazơ của phenethylamin, quy trình chiết phổ biến nhất là chiết pha rắn (SPE) với các cột SPE trao đổi cation chế độ hỗn hợp (mixed mode SPE). Trong các quy trình này, mẫu được đưa lên cột ở pH 6 hoặc 7, sau khi rửa cột SPE lần lượt bằng nước, dung dịch đệm axit và dung môi hữu cơ, quá trình rửa giải được thực hiện trong dung môi hữu cơ chứa 2% amoni hydroxit. Habrdova et al. [4] pha loãng 1 mL huyết tương trong 2 mL nước, sau khi rửa bằng nước, HCl 0,01 M và MeOH, mẫu được rửa giải bằng MeOH-NH4OH (98:2, v/v). Johnson et al. [20] và Poklis et al. [1] điều chỉnh mẫu sinh học với dung dịch đệm photphat (pH 6), sau khi rửa cột bằng nước, axit axetic 0,1 M và MeOH, quá trình rửa giải được thực hiện với 2% NH4OH trong metylen clorua (CH2Cl2)/isopropanol (IPA) (80:20, v/v). Kerrigan et al. [6] cũng điều chỉnh các mẫu bằng dung dịch đệm photphat, nhưng sử dụng một quá trình rửa kỹ lưỡng hơn với nước, axit axetic 1 M, hexane, etyl axetat và MeOH. Quá trình rửa giải được thực hiện bằng 2% NH4OH trong CH2Cl2/IPA (95:5, v/v). Poklis et al. [19] đã thực hiện một kỹ thuật SPE khác, sử dụng cột SPE đặc biệt cho phép một quy trình nhanh hơn; 0,5 mL mẫu và 0,5 mL MeOH được trộn, sau đó 300 µL được chuyển sang các cột SPE Clean Screen Fast® và hút trực tiếp vào các lọ để tiêm.
Caspar et al. [18] sử dụng hai quy trình chiết lỏng-lỏng (LLE) liên tiếp để làm sạch mẫu huyết tương. Trong đó, 1 mL huyết tương được trộn với 2 mL dung dịch natri sulfat bão hòa trong nước và 5 mL hỗn hợp dietyl ete-etyl axetat (1:1, v/v). Hỗn hợp được lắc bằng tay trong ít nhất 20 giây và ly tâm cho đến khi tách pha, và pha hữu cơ phía trên được chuyển vào bình. Tiếp theo, 0,5 mL natri hydroxit (1 M, pH 8–9) và 5 mL hỗn hợp dietyl ete-etyl axetat (1:1, v/v) được thêm vào chất lỏng còn lại và tiếp tục trộn bằng tay trong ít nhất 20 giây, ly tâm trong 2 phút. Pha dung môi phía trên được chuyển vào một bình, sau đó, được làm bay hơi đến khô. Boatto et al. [17] đã sử dụng quy trình làm sạch đơn giản hơn, bao gồm kết tủa protein (PP). Trong đó, huyết tương đã xử lý trước (1 mL) với 20 µL axit photphoric được kết tủa bằng acetonitrile.
Sau các quy trình chiết đã mô tả (SPE, LLE và PP), các mẫu đã chiết được làm bay hơi đến khô, tái hoà tan trong pha động thích hợp và được bơm lên thiết bị phân tích. Trước bước làm bay hơi, Poklis et al. [1] và Kerrigan et al. [6] khuyến nghị thêm MeOH có tính axit để giảm sự thất thoát phenethylamin trong quá trình làm bay hơi. Poklis et al. [1] quan sát thấy sự mất ổn định của cả 2CC-NBOMe và 25I-NBOMe nếu dung môi rửa giải SPE có tính kiềm được làm bay hơi đến khô. Việc thêm HCl metanolic và nước vào dung môi rửa giải kết hợp với sự bay hơi hạn chế của dung môi cho phép hiệu suất thu hồi ở mức chấp nhận được. Kerrigan et al. [6] đã nghiên cứu hiệu ứng "salting out" trước khi bay hơi để xác định sự thay đổi của mẫu có thể xảy ra do tính dễ bay hơi của một số loại thuốc ở dạng bazơ (không tích điện) hay không. Kết quả là các hợp chất 2C, 2C-T và DO bị bay hơi ở dạng bazơ (không tích điện) thấp hơn đáng kể (19–75%), so với những chất được xử lý bằng metanol có tính axit trước khi bay hơi. Tuy nhiên, Caspar et al. [18] và Habrdova et al. [4] không thực hiện bước “salting out” và không axit hóa các chất chiết trước khi bay hơi.
2.6 Dẫn xuất hóa các phenethylamin
Trong số các nghiên trong Bảng 2, chỉ có Habrdova et al. [4] thực hiện phân tích bằng GC-MS, và cần phải thực hiện bước dẫn xuất hóa. Thuốc dẫn xuất hóa đã chọn là heptafluorobutyric anhydride (HFBA), 20 µL HFBA được thêm vào mẫu và vi sóng ở 450 W trong 5 phút. Sau khi dẫn xuất hóa, các chất chiết được trộn với hexane và Na3PO4 0,5 M và 1 µL của phần hữu cơ được bơm vào hệ GC-MS. Tuy nhiên độ tái lặp đối với quá trình dẫn xuất hóa của 2C-T-2 và 2C-T-7 không tốt (có thể) do nhóm sulfur trong hai thuốc này.
2.7 Phân tách sắc ký cho các phenethylamin
Trong phương pháp GC, Habrdova et al. [4] đã sử dụng một cột mao quản không phân cực, HP-5MS (30 m × 0,25 mm i.d.) với thời gian chạy là 10 phút. Với các phương pháp LC, quá trình tách sắc ký được thực hiện với các cột pha đảo. Các pha động được sử dụng ở chế độ gradient với hỗn hợp nước với các chất điều chỉnh như axit formic và amoni axetat hoặc formate và dung môi hữu cơ, chủ yếu là acetonitril. Caspar et al. [18] sử dụng một cột Accucore Phenyl Hexyl LC (100 mm × 2,1 mm, 2,6 µm) với pha động gồm amoni formate 2 mM trong nước ở pH 3,4 chứa 0,1% axit formic (pha động A) và metanol/acetonitrile (50:50, v/v) chứa 0,1% axit formic (pha động B). Tổng thời gian chạy là 15 phút và thời gian lưu (RT) của các hợp chất 2C là 1,7–4,7 phút và các hợp chất NBOMe RT là 7,1–10,2 phút. Johnson et al. [20] sử dụng cột Phenomenex Kinetex PFP (50 × 2,1 mm, 1,7 um), với pha động là amoni formate 2 mM chứa 0,2% axit formic và acetonitrile chứa 0,1% axit formic. Tổng thời gian chạy là 4,51 phút, các hợp chất NBOMe rửa giải trong khoảng 1,5–3 phút. Poklis et al. sử dụng cột Phenomenex Luna C8(2) (100 × 2 mm, 3 µm) [1] hoặc cột Restek Allure Biphenyl (100 × 3,2 mm, 5 µm) [19]. Trong cả hai nghiên cứu, pha động là nước với amoni axetat 10 mM và 0,1% axit formic và metanol. Tổng thời gian chạy lần lượt là 10 phút (RT 3,5–5,5 phút) hoặc 13 phút (RT 7–12 phút) trên các cột tương ứng. Kerrigan et al. [6] sử dụng cột Phenomenex Luna C18 (100 × 2 mm, 5 µm), với pha động A là amoni axetat 50 mM trong nước/metanol (95:5, v/v) và pha động B là amoni axetat 50 mM trong acetonitrile/nước (90:10, v/v). Thời gian chạy của phương pháp là 6 phút cộng với thời gian cân bằng.
2.8 Phương pháp khối phổ cho các phenethylamin
Hầu hết các nghiên cứu đều sử dụng LC-MS và phương pháp phát hiện là khối phổ ghép đôi độ phân giải thấp (low resolution tandem MS) ở chế độ giám sát phản ứng đa (MRM). Hai chuyển tiếp MRM được ghi tín hiệu cho mỗi chất phân tích, các chất đều được được ion hóa bằng kỹ thuật ESI ở chế độ dương [1, 6, 19, 20]. Boatto et al. [17] đã sử dụng một thiết bị MS đơn và phân tử proton hóa (M+H+) được chọn làm ion định lượng. Caspar et al. [18] sử dụng khối phổ độ phân giải cao (HRMS). Trong đó, tác giả đã theo dõi số khối chính xác của dạng M+H+ của chất phân tích ở chế độ quét toàn dải phổ và sử dụng ít nhất hai ion mảnh đặc trưng với độ chính xác khối < 5 ppm với tỷ lệ tín hiệu trên nhiễu là 3:1 thu được ở chế độ phân mảnh tất cả ion (AIF). Các tác giả đã xác định các ion phân mảnh chỉ thị cho các nhóm, chẳng hạn như m/z 121,0653 cho NBOMe, m/z 188,1439 của các dẫn xuất fentanyl hoặc m/z 223,1235 cho các dẫn xuất LSD. Harbdova et al. [4] đã sử dụng GC-EI-MS, trong đó phương pháp MS hoạt động ở chế độ SIM, theo dõi 3 giá trị m/z cho mỗi hợp chất. Phương pháp này cũng được sử dụng như một phương pháp sàng lọc cho các dẫn xuất phenethylamin khác bằng cách theo dõi các ion mảnh đặc trưng, gồm: m/z 181, 190, 244, 290, 391, 405, 419, 437 và 451.
3 Xác định các tryptamine trong máu và nước tiểu
Bảng 3 tóm tắt các đặc điểm chính của các phương pháp phân tích được chọn để xác định tryptamines trong huyết tương, huyết thanh và nước tiểu. Tất cả các phương pháp này đều cho thấy độ nhạy, độ đặc hiệu cao và đã được thẩm định đầy đủ, một số phương pháp đã được áp dụng cho các mẫu thực. Phương pháp toàn diện nhất được công bố bởi Meyer et al. [28] cho phân tích 37 tryptamines. Caspar et al. [18] sử dụng phương pháp đã được mô tả trong Bảng 2 để phân tích nhiều dẫn xuất liên quan tới LSD, LSZ, AL-LAD và 1P-LSD.
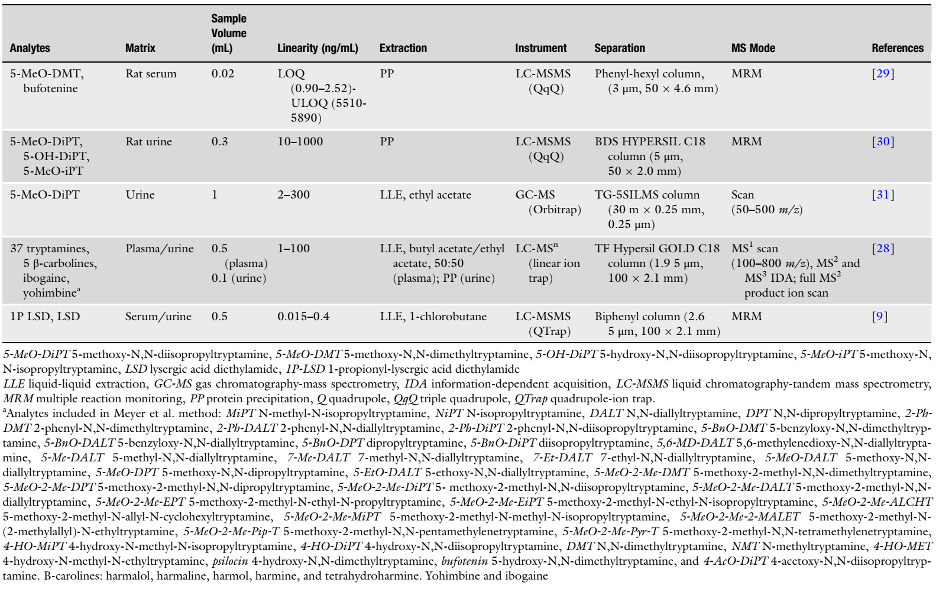
Bảng 3. Các phương pháp phân tích xác định tryptamine trong huyết tương, huyết thanh và nước tiểu
3.1 Các chất phân tích mục tiêu cho các tryptamine
Chất phân tích mục tiêu được khuyến nghị khi phân tích các tryptamine trong hầu hết các nghiên cứu liệt kê ở Bảng 3 đều là hợp chất gốc; tuy nhiên, các chất chuyển hóa liên quan cũng có thể được xác định trong các mẫu nước tiểu. 5-MeO-DiPT thường được chuyển hóa thành 5-hydroxy-N,N-diisopropyltryptamine (5-OH-DiPT) và 5-methoxy-N-diisopropyltryptamine (5-MeO-iPT) ở người [31, 32]. Mặc dù hydroxy-tryptamines, các chất chuyển hóa hydroxy và các chất chuyển hoá liên hợp khác đã được báo cáo tuy nhiên không có nghiên cứu nào được trích dẫn trong Bảng 3 thực hiện quy trình thủy phân trong quá trình chuẩn bị mẫu. Pichini et al. [21] quan sát thấy sự gia tăng nồng độ của 4-hydroxy-N,N-diisopropyltryptamine (4-HO-DiPT) một tryptamine tổng hợp sau khi thủy phân (Helix pomatia, 16 giờ, 37°C). Trong nghiên cứu này, tác giả cũng báo cáo không có sự thay đổi đáng kể nào xảy ra với 4-acetoxy-N,N-diisopropyltryptamine (4-acetoxy-DiPT) sau khi thủy phân các mẫu thực. Với các dẫn xuất mới của LSD, 1P-LSD, Grumann et al. [9] không phát hiện 1P-LSD trong cả nước tiểu và mẫu huyết thanh của người sử dụng ma túy, kết quả này hỗ trợ giả thiết 1P-LSD có thể đóng vai trò như một tiền chất của LSD in vivo [33].
3.2 Liều lượng và nồng độ của các tryptamine
Do liều lượng thấp của các chất này, nồng độ trong mẫu sinh học được dự kiến thấp trong phạm vi pg/mL - ng/mL. Liều lượng phổ biến của LSD và một số dẫn xuất là 50–200 µg [34], trong khi 5-MeO-DiPT ở mức cao hơn 6–12 mg [35]. Nồng độ trong huyết tương được báo cáo sau khi uống các hợp chất tryptamine là từ 6,2–115 ng/mL [36]. Yan et al. [31] đã phân tích các mẫu nước tiểu từ những người sử dụng 5-MeO-DiPT và báo cáo nồng độ dao động từ 1–2,8 ng/mL.
3.3 Độ ổn định của các tryptamine
Độ ổn định trong các mẫu tăng cường (fortified sample – thường là chất chuẩn của chất phân tích được thêm vào một nền mẫu thực) được nghiên cứu cho 5-MeO-DiPT và các chất chuyển hóa. Jin et al. [30] báo cáo 5-MeO-DiTP, 5-OH-DiPT và 5-MeO-iPT ổn định trong nước tiểu chuột sau 6 giờ ở nhiệt độ phòng, 14 ngày ở -70°C và sau ba chu kỳ đông lạnh-rã đông. Tuy nhiên, Yan et al. [31] báo cáo nồng độ 5-MeO-DiPT trong nước tiểu giảm 22,8–38,2% sau khi các mẫu được bảo quản trong 24 giờ ở 25°C và 5 và 7 ngày ở 4°C. Theo khuyến nghị chung, các mẫu nên được bảo quản ở -20°C cho đến khi phân tích. Với 1P-LSD, Grumann et al. [9] báo cáo rằng các mẫu huyết thanh và nước tiểu có 1P-LSD có thể được bảo quản ở nhiệt độ phòng, 5°C hoặc -20°C trong 3–5 ngày có hoặc không cần tránh ánh sáng và có thể trải qua ít nhất hai chu kỳ đông lạnh-rã đông mà không có sự suy giảm đáng kể. Tuy nhiên, sự thủy phân 1P-LSD thành LSD trong các mẫu đã được báo cáo (lên đến 21% và 4% trong huyết thanh và nước tiểu) sau 3–5 ngày bảo quản ở nhiệt độ phòng hoặc 5°C, kể cả khi tránh ánh sáng. Các tác giả khuyến cáo rằng các mẫu nước tiểu và huyết thanh nên được bảo quản ở -20°C và không cần bảo quản trong điều kiện tối. Ngoài ra, các tác giả cũng quan sát thấy rằng việc thêm NaF vào mẫu huyết thanh giúp ngăn chặn quá trình thủy phân enzym thành LSD.
3.4 Chất nội chuẩn sử dụng trong phân tích các tryptamine
Trong hầu hết các nghiên cứu liệt kê ở Bảng 3, các chất nội chuẩn được sử dụng là các chất tương tự được deuteri hóa của một số hợp chất mục tiêu của phương pháp. Grumann et al. [9] sử dụng LSD-d3, Jin et al. [30] sử dụng 5-MeO-DiPT-d4, 5-OH-DiPT-d4 và 5-MeO-iPT-d4. Meyer et al. [28] sử dụng ba tryptamine được deuteri hóa là 5-ethoxy-N-allyl-N-cyclohexyltryptamine (5-EtO-ALCHT-d4), N, N-diisopropyltryptamine (DiPT-d4), 5-ethoxy-N, N-diallyltryptamine (5-EtO-DALT-d4), citalopram-d6, trimipramin-d3 và zolpidem-d6. Shen et al. [29] sử dụng 5-Methyl-N, N-dimethyltryptamine và Yan et al. [31] sử dụng skf-525.
3.5 Quy trình chiết các tryptamine
Về quy trình chiết/làm sạch mẫu, 3 tác giả sử dụng kết tủa protein (PP) và 3 tác giả sử dụng chiết lỏng-lỏng (LLE). PP đã được sử dụng cho các mẫu huyết tương và nước tiểu. Shen et al. [29] phân tích 5-MeO-DMT và bufotenine trong 20 µL huyết tương bằng cách thêm 60 µL acetonitrile. Sau khi ly tâm, mẫu được tiêm trực tiếp vào LC-MS/MS bỏ qua bước bay hơi làm khô mẫu và tái hoá tan. Jin et al. [30] và Meyer et al. (2014) [28] thực hiện kết tủa protein trong các mẫu nước tiểu (300 µL nước tiểu và 600 µL acetonitrile hoặc 100 µL nước tiểu và 500 µL acetonitrile, tương ứng). Trong cả hai phương pháp, phần dịch nổi phía trên được làm bay hơi đến khô và tái hoà tan trước khi tiêm vào thiết bị.
Trong quy trình LLE, pha nước có pH kiềm (từ pH 7,4 đến > 12) và các dung môi hữu cơ được sử dụng là 1-chlorobutane, butyl acetate/ethyl acetate (50:50, v/v) hoặc ethyl acetate. Grumann et al. [9] đã phân tích 1P-LSD trong 0,5 mL mẫu. Tác giả thêm 1 mL dung dịch đệm borat pH 9 và 2 mL 1-chlorobutane. Meyer et al. [28] chiết 37 tryptamines từ 0,5 mL mẫu huyết tương sau khi điều chỉnh pH đến 7,4 bằng dung dịch đệm phosphate 10 mM. Dung môi hữu cơ được sử dụng là 600 µL butyl acetate/ethyl acetate (50:50, v/v). Yan et al. [31] điều chỉnh pH > 12 cho 1 mL nước tiểu sử dụng dung dịch NaOH 10% và việc chiết LLE được thực hiện với 3 mL ethyl acetate.
3.6 Tách sắc ký cho các tryptamine
Hầu hết các phương pháp phân tích đều sử dụng LC và chỉ có một phương pháp sử dụng GC. Phương pháp GC [31] được báo cáo không cần dẫn xuất hóa và cột được sử dụng là cột Thermo Fisher Scientific TG-5SILMS 30 m × 0,25 mm I.D., 0,25 µm. Việc tách sắc ký trong các phuwong pháp LC đều sử dụng cột pha đảo. Grumann et al. [9] đã sử dụng cột Phenomenex biphenyl (100 × 2,1 mm, 2,6 µm) với pha động A gồm nước khử ion với 1% acetonitrile, 0,1% axit formic và amoni formate 2 mM, pha động B gồm 0,1% axit formic và amoni formate 2 mM trong acetonitrile. Tổng thời gian chạy của phương pháp là 22 phút. Shen et al. [29] sử dụng cột Phenomenex phenyl-hexyl (50 × 4,6 mm, 3 µm) với pha động gồm 0,1% axit formic trong nước (A) và 0,1% axit formic trong metanol (B). Tổng thời gian chạy của phương pháp là 9 phút. Jin et al. [30] thực hiện quá trình tách sắc ký trên cột BDS Hypersil C18 (50 × 2,1 mm, 5 µm; Thermo Fisher Scientific Inc.) với các pha động là 0,1% axit formic (A) và metanol (B). Tổng thời gian chạy chỉ là 6 phút. Đối với phương pháp toàn diện nhất được phát triển bởi Meyer et al. [28], cột được sử dụng là cột TF Hypersil GOLD C18 (100 × 2,1 mm, 1,9 µm) với pha động là amoni formate 10 mM trong nước với 0,1% axit formic pH 3,4 (A) và acetonitrile với 0,1% axit formic (B). Thời gian chạy của phương pháp này là 25 phút.
3.7 Phương pháp khối phổ cho các tryptamine
Trong các phương pháp khối phổ để phân tích các trytamine phần lớn đều sử dụng LC-MS/MS (QTrap hoặc QqQ), dữ liệu được thu thập ở chế độ ESI dương với kỹ thuật ghi tín hiệu theo dõi đa phản ứng (MRM). Grumann et al. [9] theo dõi hai chuyển tiếp (transition) cho mỗi hợp chất; tuy nhiên, Shen et al. [29] và Jin et al. [30] chỉ theo dõi một chuyển tiếp cho mỗi hợp chất. Meyer et al. [28] sử dụng 1 phương pháp MS phức tạp hơn với thiết bị khối phổ bẫy ion tuyến tính (Qq-LIT) ở chế độ ESI dương cho việc định lượng trong huyết tương và sàng lọc trong mẫu nước tiểu các chất quan tâm. Với mẫu nước tiểu, tác giả đã sử dụng LIT để thu thập dữ liệu ở cả chế độ quét toàn dải phổ (giai đoạn MS1) và dữ liệu phổ MSn (MS2 và MS3) của các ion tiền chất sử dụng kỹ thuật ghi tín hiệu phụ thuộc thông tin cho trước (IDA, trong đó các ion tiền chất được tự động chọn từ phổ MS1, phá mảnh và ghi lại toàn dải phổ MS2 và MS3). Các chất phân tích được xác định nhờ việc tham chiếu phổ MS2 và MS3 toàn dải thu được với phổ của chất trong thư viện. Với mẫu huyết tương, thay vì sử dụng IDA, tác giả ghi tín hiệu ion sản phẩm MS2 toàn dải (PIS) của các chất phân tích mục tiêu và các chất nội chuẩn, sau đó sử dụng một ion phân mảnh đặc trưng cho từng chất để định lượng. Các chất phân tích trong huyết tương được xác định dựa trên phổ PIS của các hợp chất mục tiêu và kết quả tham chiếu với thư viện phổ sử dụng cùng các thiết lập đã được dùng cho các mẫu nước tiểu. Yan et al. [31] sử dụng GC với ion hoá EI kết hợp với khối phổ độ phân giải cao (Orbitrap). Dữ liệu được thu thập ở chế độ quét toàn dải với ở chế độ dương và theo dõi cùng lúc 3 giá trị m/z đặc trưng cho mỗi hợp chất.
4 Xác định các piperazine trong máu và nước tiểu
Bảng 4 tóm tắt các đặc điểm chính của các phương pháp được chọn để xác định piperazines trong máu toàn phần, huyết tương và nước tiểu. Tất cả các phương pháp này đều thể hiện độ nhạy, độ đặc hiệu cao và đã được thẩm định đầy đủ. Một số phương pháp đã được áp dụng cho các mẫu thực.
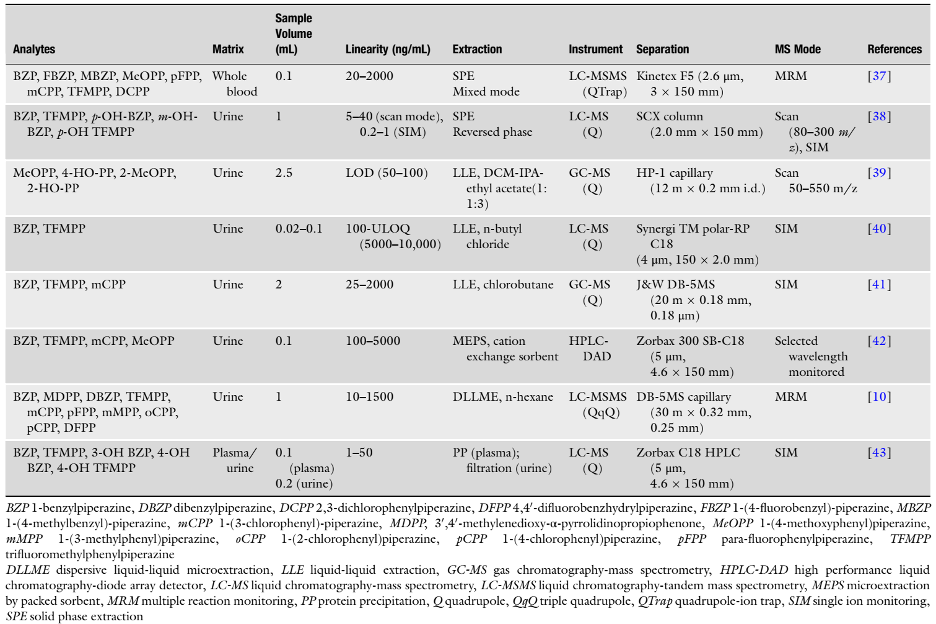
Bảng 4. Các phương pháp phân tích piperazine trong máu toàn phần, huyết tương và nước tiểu
4.1 Các chất phân tích mục tiêu của các piperazine
Hầu hết các phương pháp để phân tích cho mẫu nước tiểu tập trung vào phân tích các ion tiền chất của chất quan tâm. Tuy nhiên, các chất chuyển hóa của BZP, TFMPP và MeOPP cũng được phân tích trong nhiều nghiên cứu [38, 39, 43]. Sự thủy phân của các mẫu nước tiểu để tăng khả năng phát hiện các chất chuyển hóa dạng hydroxyl của BZP và TFMPP có thể được thực hiện bằng việc thêm enzyme phù hợp trong 12 giờ ở 37°C [38, 43]. Staack et al. [39] cũng báo cáo việc thủy phân hiệu quả các đối với các chất chuyển hóa dạng hydroxyl của MeOPP sử dụng axit, với ưu điểm chính là về chi phí.
Mặc dù việc phân tích các chất chuyển hóa tương ứng được khuyến cáo để mở rộng khả năng phát hiện và tăng thêm thông tin có giá trị trong việc giải thích một số trường hợp, nhưng điều quan trọng là các thuốc tiền chất phải luôn được phân tích cùng để hạn chế tình trạng dương tính giả [39]. Một số loại thuốc liên quan đến piperazine nhất định, chẳng hạn như thuốc ho dropropizine và thuốc chống loạn thần oxypertine, cũng có các chất chuyển hóa tương tự như MeOPP, 1-(4-hydroxyphenyl)piperazine (4-OH-PP). Do đó, việc phân tích các thuốc tiền chất cho phép làm rõ nguồn gốc của chất chuyển hóa chung này. Cũng cần cân nhắc rằng piperazine mCPP là một thuốc piperazine và cũng là một chất chuyển hóa của thuốc chống trầm cảm trazodone và nefazodone [44]. Việc phân tích đồng thời các thuốc chống trầm cảm này có thể tránh được những kết luận sai về mCPP.
4.2 Liều lượng và nồng độ của các piperazine
Piperazine được tiêu thụ ở liều lượng cao hơn phenethylamines và tryptamines dẫn tới nồng độ cao hơn trong các mẫu sinh học. Liều lượng trung bình có thể dao động từ 75–150 mg đối với BZP và 25–100 mg đối với TFMPP, tương tự như liều của MDMA [40]. BZP và TFMPP là các thành phần hoạt tính thường gặp nhất của “thuốc lắc” hoặc “cỏ dược”. 2 chất này có thể tồn tại ở các tỷ lệ khác nhau trong thuốc và có các tên gọi khác nhau, như “Charge” (50 mg BZP và 200 mg TFMPP) hoặc “Bliss” (100 mg BZP và 50 mg TFMPP) [43]. BZP và TFMPP thường được sử dụng cùng nhau là do có tác dụng hiệp đồng làm tăng đáng kể mức độ dopamine và serotonin của người sử dụng [45].
4.3 Độ ổn định của các piperazine
Các nghiên cứu về độ ổn định ngắn hạn của các piperazine trong các mẫu nước tiểu tăng cường không cho thấy sự phân huỷ đáng kể. Antia et al. [43] đã nghiên cứu độ ổn định của BZP, TFMPP cùng các chất chuyển hóa dạng hydroxyl liên quan trong các mẫu tăng cường ở nhiệt độ phòng trong 24 giờ, đông lạnh-rã đông và ở -20°C trong 3 tháng. Tương tự, Moreno et al. [42] không phát hiện sự thay đổi đáng kể nào đối với MeOPP, BZP, mCPP và TFMPP trong nước tiểu tăng cường 24 giờ ở nhiệt độ phòng. Lau et al. [37] nghiên cứu độ ổn định của tám piperazine trong mẫu máu toàn phần. Về cơ bản, benzylpiperazines ổn định hơn phenylpiperazines trong tất cả các điều kiện bảo quản, trong đó MBZP còn lại hơn 70% sau 12 tháng bảo quản ở -20°C. Trong các piperazine, MeOPP là chất ít ổn định nhất, không thể được phát hiện ở nhiệt độ phòng và nhiệt độ thấp sau 6 tháng. Do đó, các mẫu sinh học để phân tích piperazine được khuyến cáo nên được bảo quản ở -20°C cho đến khi phân tích.
4.4 Chất nội chuẩn của các piperazine
Hầu hết các nghiên cứu đều sử dụng một dẫn xuất của piperazine, như 1-(2-chlorophenyl)-piperazine [42], 1-(4-chlorophenyl)-piperazine [38], 1-(3-chlorophenyl)-piperazine [40] hoặc 1-(2-methoxyphenyl)piperazine [10] làm chất nội chuẩn. Dickson et al. [41] và Lau et al. [37] sử dụng các chất tương tự được deuteri hóa của một số piperazine như BZP-d7, mCPP-d8, TFMPP-d4. Mặc dù nghiên cứu từ Antia et al. [43] không sử dụng bất kỳ chất nội chuẩn nào và thực hiện hiệu chuẩn bên ngoài (external calibration), nhưng cần lưu ý rằng cách tiếp này không được khuyến nghị trong độc chất học lâm sàng và pháp y do sự phức tạp của nền mẫu cũng như tính bất ổn định của nhiều chất quan tâm.
4.5 Quy trình chiết các piperazine
Liên quan tới quy trình chiết trong huyết tương, Antia et al. [43] chỉ sử dụng kết tủa protein đơn giản, trong đó 100 µL huyết tương đã được loại protein bằng cách thêm ZnSO4 (20 µL, 35%) và metanol (100 µL). Sau khi trộn đều và ly tâm, phần dịch nổi phía trên được phân tích trực tiếp. Với mẫu máu toàn phần, Lau et al. [37] thực hiện chiết với cột SPE trao đổi cation chế độ hỗn hợp. Khoảng 100 µL máu toàn phần được điều chỉnh ở pH 6 và nạp vào các cột SPE. Cột sau đó được rửa lần lượt bằng nước, HCl 0,1 N và metanol, cuối cùng quá trình rửa giải được thực hiện bằng cách thêm 20% 2-propanol, 3% amoni hydroxit đậm đặc và 77% metylen clorua. Sau khi bay hơi và tái hoà tan, các mẫu được phân tích bằng LC-MS/MS.
Với các mẫu nước tiểu, các tác giả đã báo cáo sử dụng nhiều quy trình bao gồm lọc đơn giản [43], LLE [39–41], SPE [38] và các quy trình vi chiết [10, 42]. Trong tất cả các quy trình LLE, pH của mẫu nước tiểu được điều chỉnh về điều kiện kiềm sử dụng KOH [40, 41] hoặc amoni sulfat và NaOH [39], sau đó chiết với dung môi hữu cơ, như chlorobutane [41], dichloromethane-isopropanol-ethyl acetate (1:1:3, v/v/v) [39] hoặc n-butyl chloride [40]. Tsutsumi et al. [38] sử dụng cột SPE pha đảo để chiết các chất này. Trong đó, mẫu nước tiểu đã thủy phân (pH 5) được đưa lên các cột SPE, sau đó được rửa lần lượt bằng HCl 0,1%, nước và metanol, cuối cùng được rửa giải với metanol. Trong nghiên cứu này, tác giả cũng so sánh quy trình SPE pha đảo sử dụng với SPE trao đổi cation và LLE sử dụng chloroform-2-propanol (3:1, v/v). Các quy trình SPE cho thấy độ thu hồi tốt hơn so với LLE, đặc biệt là đối với các chất chuyển hóa hydroxy. Hiệu suất thu hồi dao động từ 81% đến 109% đối với SPE pha đảo, từ 20% đến 103% đối với SPE chế độ hỗn hợp và từ 6% đến 77% đối với LLE. Piperazine cũng đã được chiết từ nước tiểu bằng các quy trình vi chiết pha rắn, MEPS (vi chiết bằng vật liệu hấp phụ được đóng gói) và UA-LDS-DLLME (chiết lỏng-lỏng phân tán bằng dung môi mật độ thấp có hỗ trợ siêu âm). Trong quy trình MEPS [42], vật liệu hấp phụ được đóng gói được sử dụng là vật liệu trao đổi cation; 0,1 mL nước tiểu được pha loãng bằng 0,1 mL nước khử ion và trộn đều. Mẫu được hút/nhả thủ công qua vật liệu hấp phụ tám lần. Vật liệu hấp phụ đã được rửa bằng 250 µL axit axetic 1% và 100 µL metanol 10% trong nước và cuối cùng được rửa giải.
4.6 Dẫn xuất hóa các piperazine
Với các phương pháp GC, các quy trình dẫn xuất hóa khác nhau đã được báo cáo. Dickson et al. [41] thực hiện quá trình dẫn xuất hóa của BZP và TFMPP bằng pentafluoropropionic anhydride (PFPA). Staack et al. [39] dẫn xuất hóa MeOPP và các chất chuyển hóa bằng quá trình acetyl hóa với hỗn hợp axit acetic anhydride-pyridine (3:2, v/v) và Zhu et al. [10] đã phân tích 10 dẫn xuất piperazine bằng GC, tuy nhiên tác giả không thực hiện bất kỳ quá trình dẫn xuất hóa nào.
4.7 Tách sắc ký các piperazine
Với tất cả các phương pháp GC đều sử dụng các cột sắc ký không phân cực tương tự cho việc phân tích piperazine (Bảng 5). Với các phương pháp LC, hầu hết việc tách sắc ký được thực hiện ở chế độ pha đảo với các cột liệt kê trong Bảng 5. Liên quan tới các pha động được sử dụng, Antia et al. [43] sử dụng dung dịch đệm amoni formate (pH 4,5, 0,01 M, pha động A) và acetonitrile (pha động B), Moreno et al. [42] sử dụng amoni formate 5 mM (pH 6,4) và metanol (55:45, v/v), Vorce et al. [40] sử dụng axit formic 0,1% trong nước và acetonitrile, Lau et al. [37] sử dụng amoni formate 2 mM với axit formic 0,2% (A) và metanol với axit formic 0,1% (B). Tất cả các phương pháp đều được tách ở chế độ gradient, ngoại trừ Moreno et al. [42] sử dụng chế độ đẳng dòng và Tsutsumi et al. [38] sử dụng cột LC trao đổi cation, một cột semi-micro SCX (2,0 mm I.D. × 150 mm; Shiseido, Tokyo, Nhật Bản) sử dụng pha động gồm amoni axetat 40 mM (pH 4) và acetonitrile (25:75, v/v).
4.8 Phân tích khối phổ và đầu dò mảng diode các piperazine
Như đã liệt kê trong Bảng 4, việc phân tích các piperazine thường được thực hiện bằng khối phổ, ngoại trừ Moreno et al. [42] sử dụng HPLC-DAD (Diode Array Detector) trong đó mỗi piperazine được theo dõi ở một bước sóng cụ thể. Trong số các phương pháp được nổi bật tại Bảng 4, ba phương pháp sử dụng LC-MS tứ cực đơn [38, 40, 43]. Antia et al. [43] và Tsutsumi et al. [38] chỉ theo dõi một m/z cho mỗi hợp chất (dạng M+H+), Vorce et al. [40] thực hiện quá trình phân mảnh do va chạm trong nguồn (in source collision-induced fragmentation) và ghi tín hiệu cho ba giá trị m/z của từng hợp chất. Lau et al. [37] sử dụng LC-MS/MS theo dõi hai chuyển tiếp MRM cho mỗi hợp chất. Dickson et al. [41] và Staack et al. [39] sử dụng GC-EI-MS và theo dõi ba giá trị m/z cho mỗi hợp chất và Zhu et al. [10] sử dụng GC-EI-MS/MS ghi tín hiệu ba chuyển tiếp MRM cho mỗi chất phân tích.
5. Các phương pháp phân tích đa mục tiêu trong máu và nước tiểu
Với mục tiêu phân tích một số nhóm NPS chỉ trong một lần phân tích, một số phòng thí nghiệm đã phát triển các phương pháp phân tích đa mục tiêu trong máu và nước tiểu. Các phương pháp này bao gồm một số phenethylamin, tryptamine và piperazine, bên cạnh các NPS khác như cathinones tổng hợp. Để bao gồm trong chương này tất cả các quy trình phân tích có sẵn cho việc phân tích các phenethylamin, tryptamine và piperazines, Bảng 5 tóm tắt các quy trình sàng lọc và xác nhận đồng thời các NPS trong máu, huyết thanh và nước tiểu. Dickson et al. [41], bên cạnh việc xác nhận piperazines đã mô tả trong Bảng 4, cũng đã thực hiện sàng lọc các tryptamine và phenethylamine cùng nhiều loại thuốc khác (tổng cộng 32 chất phân tích). Phương pháp sàng lọc tương tự như phương pháp đã mô tả trong phần trước.
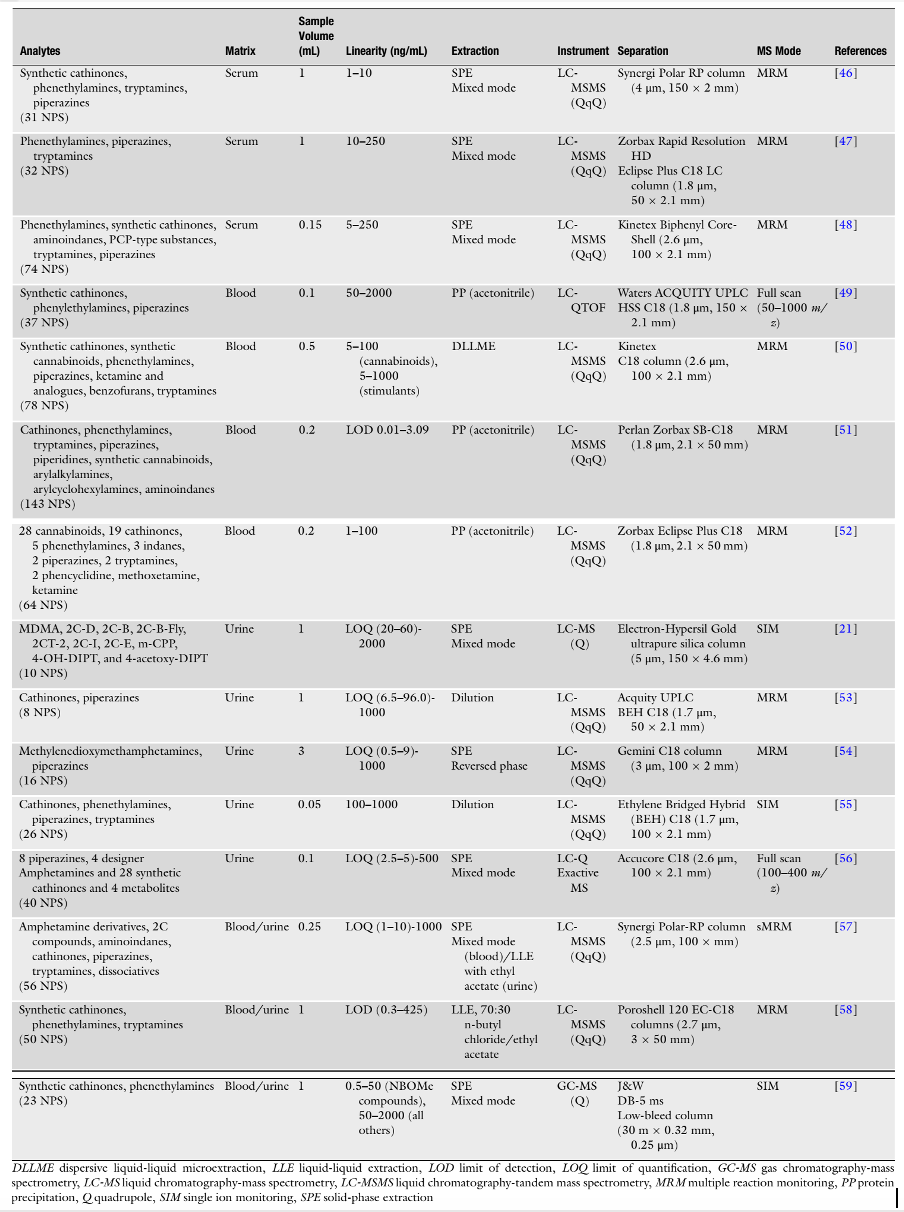
Bảng 5. Các phương pháp phân tích xác định nhiều NPS, bao gồm phenethylamin, piperazines và tryptamines trong máu, huyết thanh và nước tiểu
6. Tài liệu tham khảo
- Poklis JL, Charles J, Wolf CE, Poklis AL (2013) High-performance liquid chromatography tandem mass spectrometry method for the determination of 2CC-NBOMe and 25I-NBOMe in human serum. Biomed Chromatogr 27:1794–1800. https://doi.org/10.1002/bmc.2999
- Acuña-Castillo C, Villalobos C, Moya PR, Sáez P, Cassels BK, Huidobro-Toro JP (2002) Differences in potency and efficacy of a series of phenylisopropylamine/phenylethylamine pairs at 5-HT(2A) and 5-HT(2C) receptors. Br J Pharmacol 136:510-519. https://doi.org/10.1038/sj.bjp.0704747
- Shulgin A (1992) PIHKAL: a chemical love story. Nation Publishing Group Co. Ltd
- Habrdova V, Peters FT, Theobald DS, Maurer HH (2005) Screening for and validated quantification of phenethylamine-type designer drugs and mescaline in human blood plasma by gas chromatography/mass spectrometry. J Mass Spectrom 40:785–795. https://doi.org/10.1002/jms.853
- Braden MR, Parrish JC, Naylor JC, Nichols DE (2006) Molecular interaction of serotonin 5-HT2A receptor residues Phe339(6.51) and Phe340(6.52) with superpotent n-benzyl phenethylamine agonists. Mol Pharmacol 70:1956-1964. https://doi.org/10.1124/mol.106.028720
- Kerrigan S, Mott A, Jatzlau B, Ortiz F, Perrella L, Martin S, Bryand K (2014) Designer psychostimulants in urine by liquid chromatography-tandem mass spectrometry. J Forensic Sci 59:175–183. https://doi.org/10.1111/1556-4029.12306
- Synthetic Drug Control Act of 2011. http://www.justice.gov/ola/views-letters/112/093011-ltr-re-hr1254-synthetic-drug-control-act-2011.pdf
- 21 CFR 1308.11(d) https://www.ecfr.gov/cgi-bin/text-idx?SID=b632b274cf6322a0450af69d7c7a4f46&node=pt21.9.1308&rgn=div5#se21.9.1308_111
- Grumann C, Henkel K, Stratford A, Hermanns-Clausen M, Passie T, Brandt SD, Auwärter V (2019) Validation of an LC-MS/MS method for the quantitative analysis of 1P-LSD and its tentative metabolite LSD in fortified urine and serum samples including stability tests for 1P-LSD under different storage conditions. J Pharm Biomed Anal 174:270-276. https://doi.org/10.1016/j.jpba.2019.05.062
- Zhu B, Meng L, Cao J, Yang W, Conlan XA (2021) Simultaneous determination of 10 new
- Dean B, Stellpflug S, Burnett A, Engebretsen K (2013) 2C or not 2C: phenethylamine designer drug review. J Med Toxicol 9: 172-178. https://doi.org/10.1007/s13181-013-0295-x
- Moreira AM dos S, Oliveira HL de, Filho JFA, Florez DHA, Borges MMC, Lacerda V, Romão W, Borges KB (2019) NBOMe compounds: an overview about analytical methodologies aiming their determination in biological matrices. TrAC Trends Anal Chem Regul Ed 114:260-277. https://doi.org/10.1016/j.trac.2019.02.034
- Araújo AM, Carvalho F, Bastos MDL, Pinho PGD, Carvalho M (2015) The hallucinogenic world of tryptamines: an updated review. Arch Toxicol 89:1151–1173. https://doi.org/10.1007/s00204-015-1513-x
- Malaca S, Faro AFL, Tamborra A, Pichini S, Busardò FP, Huestis MA (2020) Toxicology and analysis of psychoactive tryptamines. Int J Mol Sci 21:9279. https://doi.org/10.3390/ijms21239279
- Monteiro M, de Bastos M, de Pinho PG, Carvalho M (2013) Update on 1-benzylpiperazine (BZP) party pills. Arch Toxicol 87:929-947. https://doi.org/10.1007/s00204-013-1057-x
- da Silva DD, Silva M, Moreira P, Martins M, Valente M, Carvalho F, de Bastos M, Carmo H (2017) In vitro hepatotoxicity of 'Legal X': the combination of 1-benzylpiperazine (BZP) and 1-(m-trifluoromethylphenyl)piperazine (TFMPP) triggers oxidative stress, mitochondrial impairment and apoptosis. Arch Toxicol 91:1413-1430. https://doi.org/10.1007/s00204-016-1777-9
- Boatto G, Nieddu M, Dessì G, Manconi P, Cerri R (2007) Determination of four thiophenethylamine designer drugs (2C-T-series) in human plasma by capillary electrophoresis with mass spectrometry detection. J Chromatogr A 1159:198-202. https://doi.org/10.1016/j.chroma.2006.10.051
- Caspar AT, Kollas AB, Maurer HH, Meyer MR (2018) Development of a quantitative approach in blood plasma for low-dosed hallucinogens and opioids using LC-high resolution mass spectrometry. Talanta 176:635-645. https://doi.org/10.1016/j.talanta.2017.08.063
- Poklis JL, Clay DJ, Poklis A (2014) High-performance liquid chromatography with tandem mass spectrometry for the determination of nine hallucinogenic 25-NBOMe designer drugs in urine specimens. J Anal Toxicol 38:113-121. https://doi.org/10.1093/jat/bku005
- Johnson RD, Botch-Jones SR, Flowers T, Lewis CA (2014) An evaluation of 25B-, 25C-, 25D-, 25H-, 25I and 25T2-NBOMe via LC-MS-MS: method validation and analyte stability. J Anal Toxicol 38:479-484. https://doi.org/10.1093/jat/bku085
- Pichini S, Pujadas M, Marchei E, Pellegrini M, Fiz J, Pacifici R, Zuccaro P, Farré M, Torre R de la (2008) Liquid chromatography-atmospheric pressure ionization electrospray mass spectrometry determination of "hallucinogenic designer drugs" in urine of consumers. J Pharm Biomed Anal 47:335-342. https://doi.org/10.1016/j.jpba.2007.12.039
- P 3rd Jacob, Shulgin AT (1994) Structure-activity relationships of the classic hallucinogens and their analogs. NIDA Res Monogr 146:74-91
- Poklis JL, Nanco CR, Troendle MM, Wolf CE, Poklis A (2014) Determination of 4-bromo-2,5-dimethoxy-N-[(2-methoxyphenyl) methyl]-benzeneethanamine (25B-NBOMe) in serum and urine by high performance liquid chromatography with tandem mass spectrometry in a case of severe intoxication. Drug Test Anal 6:764-769. https://doi.org/10.1002/dta.1522
- Sargent T, Shulgin AT, Naranjo C (1969) Structure-activity relationships of one-ring psychotomimetics. Nat Lond 221:537-541. https://doi.org/10.1038/221537a0
- Vorce SP, Sklerov JH (2004) A general screening and confirmation approach to the analysis of designer tryptamines and phenethylamines in blood and urine using GC-EI-MS and HPLC-Electrospray-MS. J Anal Toxicol 28: 407-410. https://doi.org/10.1093/jat/28.6.407
- da Cunha K, Eberlin M, Huestis M, Costa J (2019) NBOMe instability in whole blood. Forensic Toxicol 37:82–89. https://doi.org/10.1007/s11419-018-0438-5
- Curtis B, Kemp P, Harty L, Choi C, Christensen D (2003) Postmortem identification and quantitation of 2,5-dimethoxy-4-n-propylthiophenethylamine using GC-MSD and GC-NPD. J Anal Toxicol 27:493-498. https://doi.org/10.1093/jat/27.7.493
- Meyer M, Meyer M, Caspar A, Caspar A, Brandt S, Brandt S, Maurer H, Maurer H (2014) A qualitative/quantitative approach
- Shen H-W, Jiang X-L, Yu A-M (2009) Development of a LC-MS/MS method to analyze 5-methoxy-N,N-dimethyltryptamine and bufotenine, and application to pharmacokinetic study. Bioanalysis 1:87–95. https://doi.org/10.4155/bio.09.7
- Jin MJ, Jin C, Kim JY, In MK, Kwon OS, Yoo HH (2011) A quantitative method for simultaneous determination of 5-methoxy-N,N-diisopropyltryptamine and its metabolites in urine using liquid chromatography-electrospray ionization-tandem mass spectrometry. J Forensic Sci 56:1044-1048. https://doi.org/10.1111/j.1556-4029.2011.01753.x
- Yan X, Xiang P, Zhao Y, Yu Z, Yan H (2020) Determination of 5-MeO-DIPT in human urine using gas chromatography coupled with high-resolution Orbitrap mass spectrometry. J Anal Toxicol 44:461-469. https://doi.org/10.1093/jat/bkaa005
- Kamata T, Katagi M, Kamata HT, Miki A, Shima N, Zaitsu K, Nishikawa M, Tanaka E, Honda K, Tsuchihashi H (2006) Metabolism of the psychotomimetic tryptamine derivative 5-methoxy-n,n-diisopropyltryptamine in humans: identification and quantification of its urinary metabolites. Drug Metab Dispos 34:281-287. https://doi.org/10.1124/dmd.105.005835
- Brandt SD, Kavanagh PV, Westphal F, Stratford A, Elliott SP, Hoang K, Wallach J, Halberstadt AL (2016) Return of the lysergamides. Part I: analytical and behavioural characterization of 1-propionyl-d-lysergic acid diethylamide (1P-LSD). Drug Test Anal 8: 891-902. https://doi.org/10.1002/dta.1884
- Nichols DE (2004) Hallucinogens. Pharmacol Ther 101:131-181. https://doi.org/10.1016/j.pharmthera.2003.11.002
- Brandt SD, Freeman S, Fleet IA, Mcgagh P, Alder JF (2004) Analytical chemistry of synthetic routes to psychoactive tryptamines. Part I. Characterisation of the Speeter and Anthony synthetic route to 5-methoxy-N,N-diisopropyltryptamine using ESI-MS-MS and ESI-TOF-MS. Analyst 129:1047-1057. https://doi.org/10.1039/b407239c
- Callaway JC, Raymon LP, Hearn WL, McKenna DJ, Grob CS, Brito GS, Mash DC (1996) Quantitation of N,N-Dimethyltryptamine and Harmala Alkaloids
- Lau T, LeBlanc R, Botch-Jones S (2018) Stability of synthetic piperazines in human whole blood. J Anal Toxicol 42:88-98. https://doi.org/10.1093/jat/bkx090
- Tsutsumi H, Katagi M, Miki A, Shima N, Kamata T, Nishikawa M, Nakajima K, Tsuchihashi H (2005) Development of simultaneous gas chromatography-mass spectrometric and liquid chromatography-electrospray ionization mass spectrometric determination method for the new designer drugs, N-benzylpiperazine (BZP), 1-(3-trifluoromethylphenyl)piperazine (TFMPP) and their main metabolites in urine. J Chromatogr B Analyt Technol Biomed Life Sci 819:315-322. https://doi.org/10.1016/j.jchromb.2005.02.016
- Staack RF, Maurer HH (2003) Toxicological detection of the new designer drug 1-(4-methoxyphenyl)piperazine and its metabolites in urine and differentiation from an intake of structurally related medicaments using gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 798:333-342. https://doi.org/10.1016/j.jchromb.2003.10.004
- Vorce SP, Holler JM, Levine B, Past MR (2008) Detection of 1-benzylpiperazine and 1-(3-trifluoromethylphenyl)-piperazine in urine analysis specimens using GC-MS and LC-ESI-MS. J Anal Toxicol 32:444-450. https://doi.org/10.1093/jat/32.6.444
- Dickson AJ, Vorce SP, Holler JM, Lyons TP (2010) Detection of 1-benzylpiperazine, 1-(3-trifluoromethylphenyl)-piperazine, and 1-(3-chlorophenyl)-piperazine in 3,4-methylenedioxymethamphetamine-positive urine samples. J Anal Toxicol 34:464–469. https://doi.org/10.1093/jat/34.8.464
- Moreno IED, da Fonseca BM, Barroso M, Costa S, Queiroz JA, Gallardo E (2012) Determination of piperazine-type stimulants in human urine by means of microextraction in packed sorbent and high performance liquid chromatography-diode array detection. J Pharm Biomed Anal 61:93–99. https://doi.org/10.1016/j.jpba.2011.12.004
- Antia U, Tingle MD, Russell BR (2010) Validation of an LC-MS method for the detection and quantification of BZP and TFMPP and their hydroxylated metabolites in human plasma and its application to the pharmacokinetic study of TFMPP in humans. J Forensic Sci 55:1311-1318. https://doi.org/10.1111/j.1556-4029.2010.01457.x
- Baselt RC (2004) Disposition of toxic drugs and chemicals in man, Seventh edition. Biomedical Publications, Foster City
- Baumann MH, Clark RD, Budzynski AG, Partilla JS, Blough BE, Rothman RB (2004) Effects of "Legal X" piperazine analogs on dopamine and serotonin release in rat brain. Ann NY Acad Sci 1025:189–197. https://doi.org/10.1196/annals.1316.024
- Wohlfarth A, Weinmann W, Dresen S (2010) LC-MS/MS screening method for designer amphetamines, tryptamines, and piperazines in serum. Anal Bioanal Chem 396: 2403-2414. https://doi.org/10.1007/s00216-009-3394-4
- Swortwood M, Swortwood M, Boland D, Boland D, DeCaprio A, DeCaprio A (2013) Determination of 32 cathinone derivatives and other designer drugs in serum by comprehensive LC-QQQ-MS/MS analysis. Anal Bioanal Chem 405:1383-1397. https://doi.org/10.1007/s00216-012-6548-8
- Lehmann S, Kieliba T, Beike J, Thevis M, Mercer-Chalmers-Bender K (2017) Determination of 74 new psychoactive substances in serum using automated in-line solid-phase extraction-liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 1064:124-138. https://doi.org/10.1016/j.jchromb.2017.09.003
- Pasin D, Bidny S, Fu S (2015) Analysis of new designer drugs in post-mortem blood using high-resolution mass spectrometry. J Anal Toxicol 39:163-171. https://doi.org/10.1093/jat/bku144
- Odoardi S, Fisichella M, Romolo FS, Strano-Rossi S (2015) High-throughput screening for new psychoactive substances (NPS) in whole blood by DLLME extraction and UHPLC-MS/MS analysis. J Chromatogr B Analyt Technol Biomed Life Sci 1000:57–68. https://doi.org/10.1016/j.jchromb.2015.07.007
- Adamowicz P, Tokarczyk B (2016) Simple and rapid screening procedure for 143 new psychoactive substances by liquid chromatography-tandem mass spectrometry. Drug Test Anal 8: 652-667. https://doi.org/10.1002/dta.1815
- Vaiano F, Busardò FP, Palumbo D, Kyriakou C, Fioravanti A, Catalani V, Mari F, Bertol E (2016) A novel screening method for 64 new
- Bell C, George C, Kicman AT, Traynor A (2011) Development of a rapid LC-MS/MS method for direct urinalysis of designer drugs. Drug Test Anal 3:496–504. https://doi.org/10.1002/dta.306
- Montesano C, Sergi M, Moro M, Napoletano S, Romolo FS, Carlo MD, Compagnone D, Curini R (2013) Screening of methylenedioxyamphetamine- and piperazine-derived designer drugs in urine by LC-MS/MS using neutral loss and precursor ion scan. J Mass Spectrom 48:49-59. https://doi.org/10.1002/jms.3115
- Al-Saffar Y, Stephanson NN, Beck O (2013) Multicomponent LC-MS/MS screening method for detection of new psychoactive drugs, legal highs, in urine—experience from the Swedish population. J Chromatogr B Analyt Technol Biomed Life Sci 930:112-120. https://doi.org/10.1016/j.jchromb.2013.04.043
- Concheiro M, Castaneto M, Kronstrand R, Huestis MA (2015) Simultaneous determination of 40 novel psychoactive stimulants in urine by liquid chromatography-high resolution mass spectrometry and library matching. J Chromatogr A 1397:32-42. https://doi.org/10.1016/j.chroma.2015.04.002
- Ambach L, Redondo AH, König S, Angerer V, Schürch S, Weinmann W (2015) Detection and quantification of 56 new psychoactive substances in whole blood and urine by LC-MS/ MS. Bioanalysis 7:1119–1136. https://doi.org/10.4155/bio.15.48
- Fagiola M, Hahn T, Avella J (2018) Screening of novel psychoactive substances in postmortem matrices by liquid chromatography-tandem mass spectrometry (LC-MS-MS). J Anal Toxicol 42:562-569. https://doi.org/10.1093/jat/bky050
- Nisbet LA, Wylie FM, Logan BK, Scott KS (2019) Gas chromatography-mass spectrometry method for the quantitative identification of 23 new psychoactive substances in blood and urine. J Anal Toxicol 43:346-352. https://doi.org/10.1093/jat/bky109